 |
|
|
| |
New Antiretrovirals at 12th CROI
|
| |
| |
Reported for NATAP by Mike Youle, MD
Royal Free Hospital, London, UK
"...the future is rosy..."
When Richard Haubrich presented the exciting data on the use of TMC114, the new protease inhibitor from Tibotec, as a late breaker on Friday, I realised that a mere 12 years has passed since I had sat in the World Aids Conference in Berlin in 1993 with the distinct feeling that nothing was going to halt the onslaught of disease that was then engulfing my patients. Now my thoughts are like those of Dame Maggie Smith in 'The Prime of Miss Jean Brodie' who proclaims "give me a girl and she's mine for life". The vast majority of those with HIV can look forward to a long and relatively healthy life with an improving selection of agents, not necessarily needed to improve virologic control but altered to ameliorate toxicity and tolerability concerns. The biggest challenges to long lived survival with HIV are access to therapy, co-infection with hepatitis and long-term adherence, the latter often compounded by mental health and substance abuse issues. Notwithstanding the rather dubious presentation of a "new" phenomenon of rapid progression with multi-drug resistant virus from the New York crew [Abstract 973b], something that has clearly been seen in various units around the world (and reported more sensibly in the scientific press) the spectre of "super-HIV bugs" appears a relatively uncommon occurrence. More importantly, at the moment, seems the inexorable rise of sexually transmitted Hepatitis C, this co-infection being the contender for greatest increase in HIV-disease related mortality, and the emergence of lymphogranuloma venerum [Abstract 896] and multi-drug resistant Staphylococcus aureus (MRSA) [Abstract 877] infections, markers of unabated sexual health risk taking, mainly by gay men.
So what did the 12th Conference on Retroviruses and Opportunistic Infections have to offer in the way of new antiretrovirals. There was a veritable smorgasbord of agents on display. There were novel mechanisms of action such as maturation inhibitors and a nucleotide competitive reverse transcriptase inhibitor entry and overall around 20 agents were discussed that were not already licensed for use. The drugs on offer at this meeting are noted below with links to their abstract presentation at the conference in the Table below.
| |
| |
| |
 |
|
| |
| |
Many of these compounds look to have significant advantages over the drugs we currently use and some hold out significant promise for those who have exhausted their therapeutic options.
To examine the new agents in the order in which they attempt to prevent the virus from entering, integrating and disrupting the lifecycle of the cell we come first to the entry inhibitors and receptor blockers. On Wednesday at a session on chemokine receptor blockade, Stephen Harrison from Harvard took on explaining the intricacies of the structural biology of the HIV envelope which must interact with the cell surface to enable viral RNA entry. He explained that "little is known about the trimeric organisation" of this structure in the pre-cleaved state when gp41 is still bound together with gp120. Much more is understood about the process with which fusion occurs involving the 6-helix bundle which unzips, a process blocked by the first agent licensed in this extra cellular mechanism, enfurvitide or T20. It is the conformational changes that occur as the virus approaches the cell, is modified from a liganded to an unliganded form and performs a docking manoeuvre which allows for the action of these new drugs. Much of the work requires the elucidation of the crystal structure of the molecule in various states and at the present can only be imputed from the action of blocking agents. Lin and co-workers showed data on BMS-488043 a small molecule which attaches to gp120, causing a conformation change which disrupts the linkage to the CD4 receptor [Abstract 544]. Resistance to this agent has been mapped to a deep pocket in the inner domain of the protein.
Moving on from the structural complexity of this extra-cellular dance Donald Mosiers, from the Scripps Institute in La Jolla, examined the pre-clinical development of chemokine receptor inhibitors. He first noted that at least CCR5 offers an ideal target against HIV since it is well conserved, necessary from transmission and has evidence that reduced activity is protective against HIV infection and progression (_CCR5 homo- and heterozygotes respectively). He then broke the potential drugs into three groups:
1. Large molecules: e.g. PRO 140 (Progenics),
2. Intermediate size molecules: such as modified native ligands (Met-RANTES, AOP-RANTES, NNY-RANTES, etc) which render CCR5 inaccessible,
3. Small molecules: against CXCR4 (AMD3100) or CCR5 (TAK-652, UK427,857, SCH C/D, CMPD167, GW873140, UCB35625 etc) which interpolate into the membrane binding domain.
However, many of these agents show a wide range of sensitivities against HIV-1 isolates of up to 2 log10 which will cause challenges in their development. We may for the first time be presented with therapies with widely differing levels of activity within individuals. With CCR5 coding region variability and CCR5 promoter polymorphisms amongst the reasons for variable response we may find these inhibitors effective at distinct stages of HIV disease. Host genetics may also have a marked influence, something that is just being recognized for other agents we have used for many years such as nevirapine [Abstract 650]. On the positive side it seems that many mutations are required to block R5 or X4 virus and escape from co-receptor blocking by receptor switching may be rare. In addition work in both SCHID-HU mice and primates suggests that these may be fruitful agents in the search for safe effective microbicides.
Next Dan Kuritzkes of Harvard Medical School walked us through the clinical activity and early efficacy studies of the drugs which had reached clinical studies. He first noted that the development of the only CXCR4 blocking agent, AMD3100, had recently been stopped and that it was not clear whether blocking this co-receptor was safe since in mice total blockade appears lethal. Moving on he presented potted histories of the three agents furthest along in development.
Schering D (417690) has a molecular weight of 650 making it, as the other relatively small, and therefore hopefully easy and cheap to make; has a long half-life, boosted by ritonavir, and synergy with existing compounds. Data was presented at last years meeting of a 1.5log drop in HIV RNA after 10 days of monotherapy, with emergence of one mixed population (R5/X4) individual who reverted to R5 virus by day 14. This drug has just started large scale efficacy studies.
UK 427,857 from Pfizer has finally been named as Maraviroc and has been dosed in around 400 individuals to up to 900mg, although above 600mg some postural hypotension occurs which may limit further dose escalation. Drug levels are reduced by 50% when food is co-administered and there are interactions with several other antiretrovirals induced by CYP3A4 interactions. A novel probe study was presented by Muirhead and co-workers [Abstract 663] which showed efavirenz to result in 50% reduction in Maraviroc levels whilst a doubling of drug exposure occurred when it was added to Kaletra containing regimens. Nevirapine, meanwhile, showed a marginal increase in Cmax but no effect on AUC12. These data underline the potential dosing challenges as we move this group of drugs into the marketplace. Once again at 10 days around 1.5log drops are seen in HIV RNA. Within the first studies one patient switched receptor usage from R5 to X4 virus with unclear reasons or implications. Kuritzkes noted that "the current sensitivity of tropism assays could be called into question..." and that the use or these agents could result in emergence of pre-existing minority variants due to selective pressure against R5 virus. Finally it appears that subtly changing the structure of a series of CCR5 antagonists allows resistance to UK427,857 to be overcome which is comforting and suggests that class resistance does not result for resistance to this agent [Abstract 96].
The third agent discussed in this talk was the Glaxo-Smith-Kline (GSK) R5 blocker GW873140, a spirodiketopiperazine, which has been given to around 40 patients at doses of 200-600mg and has a similar log drop to the other molecules mentioned above. It also appears synergistic with other R5 and X4 blockers in vitro [Abstract 543]. Once again a dual tropic patient was unmasked by the addition of this agent with X4 emerging at day 10. A study by GSK [Abstract 77] showed that after drug withdrawal significant (>50%) receptor occupancy occurs for at least 5 days, a dose dependent effect which appears to be true of the other drugs which bodes well for those patients with adherence issues. In contrast with Maraviroc this agent does not affect lopinavir/ritonavir levels but the latter causes a 7-fold increase in GW8731140 levels [Abstract 664]. These pharmacokinetic data suggest that the interaction between CCR5 antagonists and other antiretrovirals is going to be a challenge for clinical practice in the coming years and will further enhance the importance of therapeutic drug monitoring.
Two other molecules had data presented on them at the meeting, TAK-652 from Takeda and CMPD167 from Merck. TAK-652 is the third offering from this Japanese group replacing the original lead compound TAK-779 and the follow-up one,TAK-220. This version has an IC50 of 1nm and levels achieved after a single dose of 8.8nm suggesting that once daily dosing will be possible as well as an impressive range of activity against resistant isolates as well as synergy or additive effects with other antiretrovirals [Abstract 541, 542]. Finally the role of CCR5 blockers in prevention was assessed in a primate model, by Veazey and co-workers, using CMPD 167. Intravaginal CMPD 167 mixed with hydroxyl methyl cellulose was administered followed by a challenge with SIV. Four out of 5 treated animals were not infected compared to the control group where all 6 acquired infection [Abstract 128], opening the way to further work in this area.
Finally, Chris Petropoulos from Virologic gave an overview of the multi-factorial process of viral entry, each step of which has a particular pathway to drug resistance. Both competitive (T20 binding) and non-competitive resistance (utilization of inhibitor-receptor complex) occur as well as potentially receptor switch or escape (which may be CCR5 independent). Probably these assays will differ quite markedly from each other.
Once again the Dream works team from Tibotec, ably supported by the master Rudi Pauwels, now relocated to St-Lgier in Switzerland, came up with something new, a group of drugs called nucleotide-competing reverse transcriptase inhibitors (NcRTI's). The work was presented by Dirk Jochmans and focused on compound X which through a series of elegant experiments appears to differ structurally from the NRTI's and functionally from the NNRTI's, i.e. it binds the active site of RT to block DNA polymerisation and nucleotide binding [Abstract 156]. When questioned Jochmans suggested that this agent was a part of a lead optimisation programme; that it works only on lentiviruses and that studies on resistance and mitochondrial DNA were ongoing.
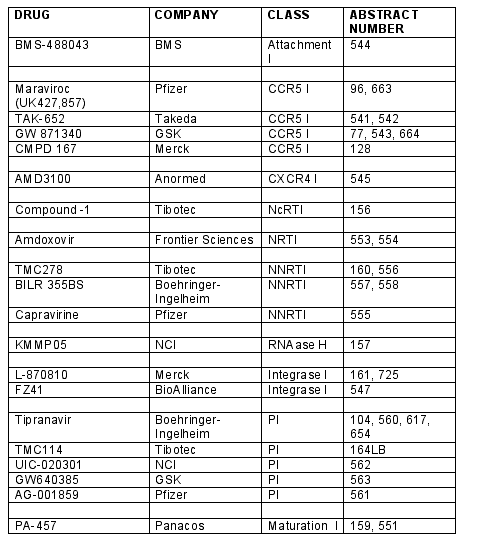
Several new non-nucleoside reverse transcriptase inhibitors were showcases at the meeting with the most exciting data being for TMC278, a diarylpyrimidine or DAPY compound. It is an extremely potent agent, EC50 of 0.5nm for wild type virus and appears to work against multiple NNRTI resistant mutants up to as many as 8 mutations, with a half-life of 38 hours and potentially active metabolites [Abstract 556].
| |
| |
| |
 |
|
| |
| |
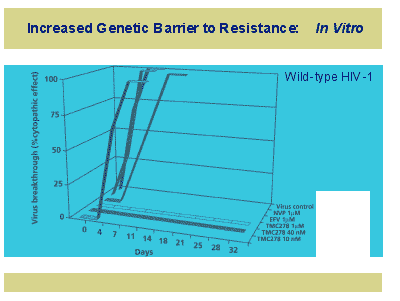
Clinical data was presented by Frank Goebel from Berlin [Abstract 160] which comprised a 7 day randomized, double blind, dose ranging monotherapy study, followed by roll-over to standard antiretroviral therapy; TMC278-C201. The study was conducted in subjects with T4 levels from 75-500 and viral loads >5,000copies/mL
| |
| |
| |
 |
|
| |
| |
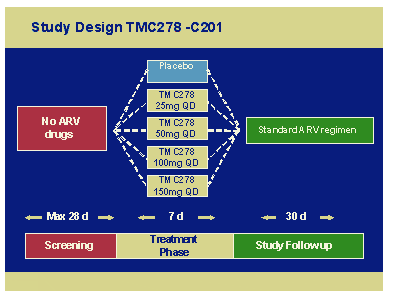
There were 9 sites in 3 countries and 47 male subjects were randomised to 25mg, 50mg, 100mg and 150mg daily for 7 days. Viral load median fall was 1.2log with no real difference across doses suggesting that much lower doses may be just as efficient.
| |
| |
| |
 |
|
| |
| |
No resistance developed and there were no pharmacokinetic or pharmacodynamic relationships seen. No significant adverse events were reported and a phase III study is due to start soon, when questioned the speaker confirmed that TMC125 and TMC 278 will both continue in development for the foreseeable future.
Boehringer-Ingelheim showed data on their new NNRTI, BILR 355BS, a novel 8-substituted dipyridodiazepinone, for the first time [Abstracts 557, 558]. The agent has good activity against wild type and resistant HIV but with a half-life of 2.5 hours it requires ritonavir boosting (100mg once daily) to achieve a half life of 16-17 hours. This may be the limiting factor in the suitability of this particular molecule to be developed.
Finally against this target two dioxolane thymine nucleosides were presented. One, the thymidine DOT showed efficacy in vitro against many of the nucleoside resistant mutations, [Abstract 554], but until further studies are undertaken it is unclear whether this agent suffers from the same toxicity profile as other thymidine analogues. The original compound DAPD or amdoxovir was evaluated in a double blind, placebo controlled, clinical study where the agent or placebo was added to optimized background which contained enfurvitide in all patients [Abstract 553]. The study halted recruitment when the development of amdoxovir was called into question. Although the drug appeared safe there was no appreciable difference between it and placebo but the study design was flawed and unlikely to produce meaningful results. The further progress of this agent is unclear at the moment.
In a session on New Targets, Michael Parniak from the University of Pittsburgh gave an elegant lecture on the potential for RNAaseH to be a suitable therapeutic target. This an endonuclease which selectively degrades the RNA strand of the double stranded RNA/DNA duplex and is both absolutely essential and specific since the cellular version resides in an alternate cellular compartment. Even so there are few inhibitors in development or even described although the N-acyl hydrazone compounds are now under evaluation in the laboratory. Daniel Himmel from Rutgers University presented data on one of the first putative RNAaseH inhibitors [Abstract 157], although, as he showed elegantly with X-ray crystallography, the active binding site for KMMP05 is at least 40A away from the active site in the palm of the P66 subunit between the primer grip and the polymerase binding site. It may be that this molecule works by deflecting the trajectory of the nucleic acid or through altered enzyme processivity. One question was asked concerning the potential for mutations within the NNRTI binding site at positions 188 or 108 to adversely affect these molecules; only time will tell.
The next target that came under scrutiny was integrase and an overview of the area was provided by the doyen of the target, Daria Hazuda from Merck Research Laboratories. Whilst providing a resume of the various types of integrase inhibitors she concentrated on the group which had moved the most, the strand-transfer agents of which several companies including GSK, Japan Tobacco, Gilead, Pfizer and BMS have potential molecules under development. At least one, L870810, has shown antiviral activity. This molecule was shown to be reduced in efficacy by mutations at positions 72, 121, 125 and 151 changes occurring within a deep binding trench [Abstract 725]. Some evidence exists of low cross resistance between agents with only the N155S causing reduced sensitivity across both the diketoacids and the naphthyridine carboxamides. Susan Little presented the only hard data at the meeting on this group of drugs which consisted of double blind placebo controlled study of L870810 (4 drug:1 placebo) as 10 days of monotherapy in 30 naïve or experienced (but off drug) subjects (Protocol 004) [Abstracts 161]. Dosing was 200mg (7 subjects) or 400mg (17 subjects) twice daily versus placebo (6 subjects) and the baseline T4 count was around 460 and viral load of 4.6log. In the higher dose group median viral load decline was 1.7log [6/16 <400copies/mL; T4 rise +89 cells (+30-+148)] and in the lower dosage arm a 1.73 log drop was seen [VL<400 1/5 subjects; T4 +73 (-107 - +253)]. Rebound occurred at varying rates over the next 24 days. This drug has been put of hold due to non-human toxicity concerns.
The other integrase inhibitor to have data shown came from BioAlliance Pharma, Paris, France [Abstract 547]. The styrylquinolones act at pre-integration step of the viral life-cycle and as such are distinct from the strand transfer inhibitors. FZ41 appears to be both synergistic, in vitro, with the latter group of integrase inhibitors as well as a range of reverse transcriptase inhibitors which makes it a particularly attractive proposition to develop.
To look at the protease inhibitors first what is really required to improve things here are agents that do not require pharmacokinetic boosting with ritonavir to achieve suitable serum levels to inhibit the prevalent viral strains and agents that do not affect lipid metabolism or affect insulin sensitivity. All of the currently licensed product appear to have lesser or greater effects on these parameters and it is this associated toxicity that most impacts on the likelihood of a patient staying on the drugs long-term. There was little at the meeting to suggest that the newer agents in current clinical development are going to be much better than what we have in these respects; however they do seem to be much more potent. The nearest agent to licensing, which is freely available across many developed countries is tipranavir from Boehringer-Ingelheim.
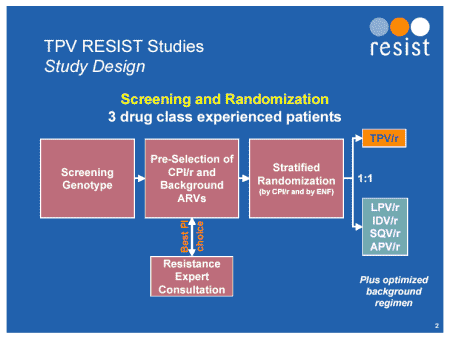
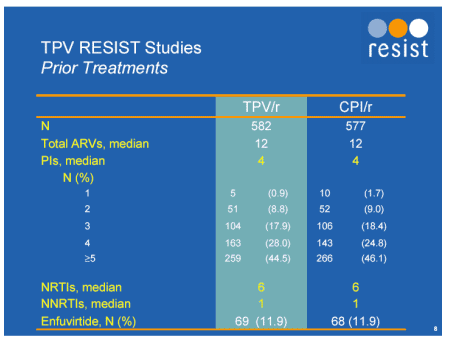
Twenty four week data was shown in two large studies of tipranavir against a comparator PI, RESIST-1 and -2 [Abstract 560]. The were phase 3, multi-centre, randomised, open-label studies of standard of care containing either tipranavir 500mg boosted with ritonavir 200mg twice daily or a comparator ritonavir boosted protease inhibitor. 1483 patients were randomised and 1159 were available for analysis at week 24. By this point viral load was less than 400copies/mL in 34% in tipranavir/r-treated individuals compared to 18% in the control arm and T4 cell rise was 31 and 6 respectively. Treatment response at 24 weeks in subjects using T20 was 58% (tipranavir/r group) compared to 26% (Comparator PI/r group).
| |
| |
| |
 |
|
| |
| |
There was heavy exposure to antiretrovirals in each arm with a median exposure to PI's of 4 although over 45% had had greater than 5. At week 24 the combined analyses showed that 41% of the tipranavir/r achieved a treatment response versus 19% of the comparator PI/r arm (P<0.0001).
| |
| |
 |
|
| |
| |
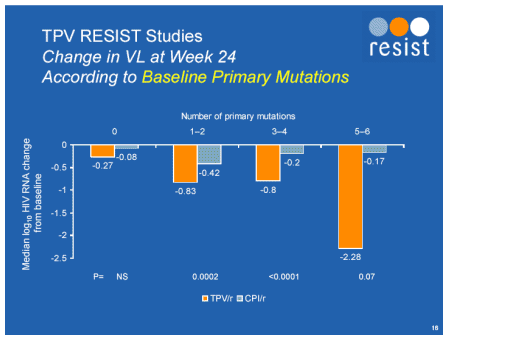
Jonathan Schapiro presented the relationship between outcome and baseline resistance analysed in a number of ways [Abstract 104]. By whichever way the resistance at baseline was characterized; all mutations, primary PI mutations, tipranavir composite score, there was a significantly better outcome for tipranavir treated subjects.
| |
| |
| |
 |
|
| |
| |
However the presence of greater than three of the mutations shown below resulted in a short lived viral load decline which had trended back to approximately 0.5log below baseline by week 24. Clearly this drug, like all other new agents does significantly better when combined with other effective antiretrovirals and if the patient has not received T20 (enfurvitide) then it would be criminal to use one without the other, especially since the pharmacokinetic interactions of tipranavir with other PI's makes their combination unfeasible.
| |
| |
 |
|
| |
| |
Albert Wu from Johns Hopkins n Baltimore resented a poster on the effect of tipranavir in the RESIST studies on Health Related Quality of Life (HRQOL) which showed that there was a significant improvement in general health and mental health scores in the tipranavir arms compared to the comparator and significant rises (P<0.05) for all dimensions of the HIV-MOS tool from baseline [Abstract 617]
Probably the most interesting data of the conference for those who treat late stage HIV disease was the protease inhibitor for Tibotec, TMC114. This has been evaluated in a two large multi-centre, international studies (TMC 114-C213/C202) in highly experienced, triple class experienced patients [Abstract 164LB].
| |
| |
 |
|
| |
| |
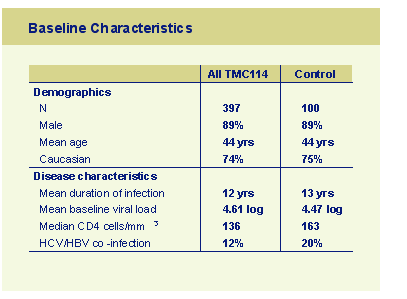
Patient disposition showed the study was mainly conducted in Caucasian males who had been heavily pre-treated with an extensive antiretroviral exposure (median 11 drugs, 17% prior exposure to T20 (12% in the control group) and 8% had used tipranavir. Each group had a median of 18 PI mutations of which 8 were known resistance mutations and 3 were primary PI-resistance mutations. Twenty nine percent of the comparator arms used double boosted PI's but less than 20% of subjects had any available PI to which they retained sensitivity on an phenotypic assay.
| |
| |
| |
 |
|
| |
| |
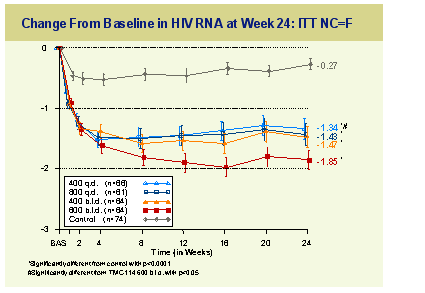
Planned interim analysis at week 24 showed significant reduction in viral load in the TMC114 arms compared to the control arm and some dose relationship with best results at the 600mg twice daily dose level.
| |
| |
 |
|
| |
| |
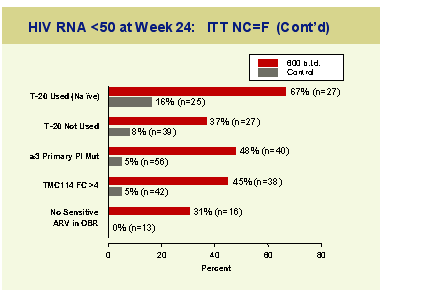
Two percent of TMC114 patients discontinued for virological failure versus 43% in the control group and the median T4 rise was 75 cells at the highest TMC dose compared to 15 cells in the control arm. A sustained mean VL reduction of 1.85log compared to 0.27 log and the proportion of subjects with a>1log reduction in viral burden was 72% versus 16%. Importantly over the study no significant changes were seen in markers of liver function or in lipid markers.
| |
| |
| |
 |
|
| |
| |
Several other early studies of novel proteases were also showcased. GSK has a drug 640385 which need to be given with ritonavir boosting and was tested in a repeat dose blinded placebo controlled study at doses of 100mg and 250mg once daily, 50mg, 150mg and 300mg twice daily always accompanied with 100mg doses of ritonavir [Abstract 563]. A further arm with 800mg of 640385 twice daily without ritonavir was also included but the latter showed AUC values of less than a fifth of the boosted regimens which all fared much better and achieved levels well above the value of 28ng/mL, calculated to be necessary to suppress PI-resistant isolates. Further studies are about to commence in PI-experienced patients.
Pfizer has a new PI, AG-001859 with reasonable data from in vitro studies against resistant isolates [Abstract 561] as have NCI in conjunction with Japanese group, UIC-02031 [Abstract 562]. This is a non-peptidic PI which incorporates cyclopentanyltetrahydrofuran and has potent activity against a wide range of multi-drug resistant HIV isolates. Both of these agents have a long way to go before reaching the clinical setting.
PA-457 is the first molecule of a new and exciting class of compounds, called maturation inhibitors, which interfere with the creation of new virions prior to them budding from the surface of the infected cell. It prevents the conversion of the HIV-1 capsid precursor, CA-SP1 (p25) to mature capsid proteins (p24) resulting in defective core condensation and release of non-infectious viral particles. David Martin from Panacos Pharmaceuticals showed data from a double blind, placebo controlled single dose study which evaluated doses of 75mg, 150mg and 250mg and placebo in 4 groups of 6 patients who were antiretroviral naïve or off therapy for at least four weeks; two subjects had extensive antiretroviral drug resistance mutations [Abstract 159]. The drug had been shown to be well tolerated in healthy volunteers, [Abstract 551], with along half-life and in this study of HIV-infected individuals; no treatment emergent side effects were noted. Viral load decline was significantly reduced (P<0.05) in a dose-dependent manner with median reduction at the highest dose of 0.51log and a greatest decline of 0.73log. Return to baseline viral RNA levels was inhibited for at least 20 days in the 250mg dose arm. Further studies are underway.
So, all in all, a fantastic show from this meeting, with lots of candidate agents being evaluated for treatment. Many of the therapies are being evaluated as single agents being given as add on or replacements compared to the best option, both being used with an optimized background regimen derived form clinical history, resistance test results and physician experience; the latter being clearly of great importance in terms of outcome. The use of single new drugs in someone with multi-resistant virus should be discouraged and trial design should reflect the new paradigm that there is a high likelihood of failure in this situation and combinations of agents to which the virus will sensitive provides the best hope for suppression.
So, in conclusion, the future is rosy and now what we must deal with is developing effective therapies for hepatitis C, where the story is much less favourable and late HIV diagnosis which remains a socio-political issue.
Thank you for those who shared slides from their presentations.
Mike Youle
| |
| |
| |
|
|
|
|
|