 |
|
|
| |
CLINICAL PHARMACOLOGY AT THE 13th CROI
|
| |
| |
New drugs (integrase, next generation fusion inhibitors), TDM, tenofovir nephrotoxicity, drug-drug interactions (TMC-125), PA-457, PK during pregnancy, Pk in sanctuary sites (CSF, gential tract), atazanavir/r monotherapy.
Feb 5-8, 2006
John G. Gerber, M.D.
Professor of Medicine and Pharmacology
Divisions of Clinical Pharmacology and Infectious Diseases
University of Colorado Health Sciences Center
Denver, Colorado
The 13th CROI was held in Denver, Colorado in the beautifully renovated downtown Convention Center. The weather in Denver was truly spectacular and there was plenty of snow in the mountains for those who combined this important meeting with some skiing time. This year the number of abstracts that dealt with clinical pharmacology issues was limited. TDM abstracts were noticeably absent which suggest to me that the need for TDM must be waning with the use of safer and more effective antiretroviral therapies. With numerous drug regimens resulting in greater than 90% efficacy in reducing HIV-RNA to <50 copies/mL in antiretroviral naive subjects, it is difficult to see how TDM can improve upon that. Perhaps TDM of protease inhibitors (PI) in the treatment of partially PI-resistant virus may turn out to be useful, but so far there are no prospective studies to demonstrate that. One interesting abstract regarding the TDM of lopinavir was abstract number 590 by Podsadecki et al. from Abbott Labs. Adherence data using MEMS caps were collected from the Abbott trial in antiretroviral naive subjects, comparing once daily Kaletra (800/200) to twice daily Kaletra (400/100) [Trial MO2-418]. In a previous presentation (ICAAC 2005, Abstract H-522) the authors showed that once daily dosing results in better drug adherence than in the twice daily dosing, but the improved adherence did not result in better outcome with once-daily Kaletra . The present analysis examined the longitudinal differences in adherence in these subjects. Interestingly the authors demonstrated that selective adherence is common and perfect adherence for a few days prior to TDM is a frequent mode of behavior. Since lopinavir has a short plasma half-life, taking the drug correctly for a few days before performing TDM can result in a therapeutic concentration of lopinavir even when the overall adherence was not good. Thus obtaining plasma concentration of lopinavir may not give an accurate and meaningful measure of overall adherence to therapy. Interestingly the opposite event, excellent adherence but missing doses prior to TDM occurred very rarely. Although we all suspected that "white coat" adherence does happen, this study demonstrated that measuring lopinavir concentrations as part of the TDM may mislead physicians about overall adherence to a drug regimen. A practical example of this may be a patient with detectable HIV-RNA despite good drug concentration with a wild-type virus.
In organizing the coverage of the Clinical Pharmacology at the 13th CROI I chose to review selected abstracts that I felt were important in the clinical scenario. I most likely missed some abstracts that are important and I want to apologize up front for that. I decided to cover clinical pharmacological aspects of new drugs, important drug-drug interactions, tenofovir nephrotoxicity, drug PK in pregnancy, and a few abstracts dealing with sanctuary site concentrations of drugs. Finally included are some miscellaneous topics.
Clinical Pharmacology of New Drugs.
Of all the new drugs presented at this meeting, the integrase inhibitors were clearly in the forefront of clinical efficacy and excellent tolerability in HIV-infected subjects harboring multi-drug resistant virus as well as in drug-sensitive virus. I will let others discuss the antiviral efficacy of these drugs but I want to concentrate on the pharmacologic differences between MK-0518 (Merck) and GS-9137 (Gilead).
MK-0518 is metabolized by UGT 1A1 glucuronidation and has to be administered twice daily for optimal efficacy. The interesting aspect of this drug is that it is not metabolized by CYP isoforms and adequate concentrations can be achieved without metabolic boosting. However the UGT 1A1 isoform that metabolizes MK-0518 is inhibited by atazanavir and it is with great anticipation we are waiting for the pharmacokinetic drug-drug interaction results of MK-0518 when combined with ATV. Especially important will be the effect of ATV on the plasma half-life of MK-0518 to see if the drug can be given once-a-day. In addition based on the ability of RTV to induce all UGT isoforms, the prediction is that RTV would reduce the exposure to MK-0518. However abstract 566 suggested that integrase inhibitors are substrates for Pgp, which would make RTV interaction more complex and difficult to predict.
GS-9137 is metabolized by CYP3A4 and is an inducer of that CYP isoform. This drug has a major interaction with RTV. RTV increases GS-9137 exposure 20-fold and prolongs the serum half-life from 3 hours to 9 hours making this a once-a-day drug. With the prolongation of t1/2 3-fold, RTV likely inhibits the systemic clearance of GS-9137, but the increase in bioavailability is likely also secondary to an effect on intestinal CYP3A as well as Pgp. Nonetheless this integrase inhibitor will likely be developed as a once-a-day drug with RTV which is an advantage, but the use of RTV will complicate the use of many concomitant drugs because of the multiple drug interactions with RTV.
Abstract 48 (from Timeris and Roche) dealt with TR-290999 and TR-291144 peptides derived from gp41HR2 region partially overlapping with the enfuvirtide (ENF) sequence. TR-290999 has an IC90 of 1 nM which is:
- 7-times more potent than enfuvirtide
- Sensitive to enfuvirtide and T-1249 resistant isolates (1 nM to 5 nM increase in IC90; ENF 7 nM to 1170 nM increase in IC90)
- 10-fold slower clearance than ENF
- 100% subcutaneous bioavailability
The plan is to develop a sustained release formulation of TR- compounds for once weekly dosing.
Gp41 mutations reduce susceptibility to fusion inhibitors but with these second generation drugs with three codon mutations there is no change in IC50 but 4 or more codon mutations there is significant reduction in susceptibility. If the drug can be administered once weekly and results in fewer local injection site reactions, it will be a major advance in the therapy of multi-drug resistance HIV.
Abstract 52 dealt with PA-457 which is a viral maturation inhibitor that works by blocking the gag processing step. The drug has t1/2 of 60-70 hours, metabolized by hepatic glucuronidation, and is 99% protein-bound. The drug has linear pharmacokinetics - t1/2 in this study was 72.8 hrs
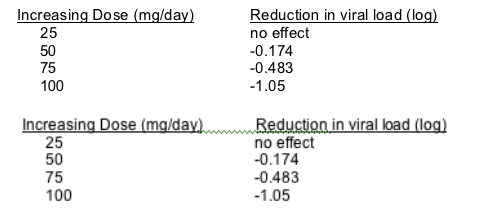
Emax model was used to evaluate the maximum effect and the maximum dose to achieve maximum effect. However the doses used were way below maximum dose and only two doses gave a truly measurable response which means that the error in the calculation of maximum dose and effect is quite high since there are only two points to the curve and neither was close to maximum. Nonetheless the calculated maximum dose was 360 mg with maximum response of - 1.6 log10. If this drug can withstand the safety issues associated with using higher doses, it may find a place in the therapy of multi-drug resistant HIV.
Pharmacokinetics during pregnancy
Abstract 707 presented by Khuong-Josses et al. followed trough concentrations of nelfinavir in pregnant subjects. NFV trough was <1mg/mL in 45% of subjects and 25/37 women had detectable viral load (>50copies/mL). However no vertical transmission was reported. The viral load tended to be lower in subjects with Cmin of NFV >1mg/mL. Overall NFV may not be the optimal drug during pregnancy because of low plasma concentration, but safety and relative efficacy is well established. Interesting part of this trial is the observation that higher dose of NFV did not increase the Cmin. A comparative study with Kaletra may be worthwhile but which end-point should be the focus, vertical transmission or development of PI resistance?
Rodman et al. reported the use of tenofovir 600 mg at the onset of labor with standard intravenous zidovudine in women who were on HAART during their pregnancy to examine the pharmacokinetics of tenofovir during delivery (abstract 708). Maternal TDF plasma concentrations were lower than reported for non-pregnant adults on this dose or the steady state TDF concentration with 300 mg/day. Cord blood concentration approximated maternal blood concentration but infants had very low plasma concentration of TDF. No toxicity was observed in this small study and there was no vertical transmission (but most had very low HIV-RNA and were taking HAART). This study basically demonstrated that infants would need to be dosed with TDF post delivery if therapeutic concentration is to be achieved. This was an interesting and important study to assess the potential use of single dose tenofovir to prevent mother to child HIV transmission in developing countries with limited resources.
Abstract 709 by Lyons et al. from St. Thomas Hospital followed 16 pregnant women on Kaletra (400/100) b.i.d. with LPV trough concentrations. 15/16 achieved trough concentrations above 1000 ng/mL (median 3660 ng/mL) which was lower than in non-pregnant women on this dose. 14/16 had HIV-RNA less than 50 copies/mL and the two with detectable viral load had adherence issues. There was no vertical transmission in this small group. These data suggest that if wild-type HIV is present the usual dose of Kaletra (400 mg LPV/100 mg of RTV) b.i.d. may be adequate for therapy during pregnancy.
Mirochnick et al reported from PACTG 1026s on the pharmacokinetics of LPV in 23 pregnant women during the 2nd trimester, 3rd trimester, and post-partum (abstract 710). Intensive PK was performed on these subjects. Doses were 400/100 b.i.d. during 2nd trimester, 533/133 b.i.d. during the third trimester and post-partum. The authors considered adequate LPV AUC >52 mg*hr/mL (10th percentile in non-pregnant women taking Kaletra ). Cord blood concentration was 25% of the maternal plasma concentration. The goal AUC was achieved in 5/8 subjects during the second trimester, 20/23 in the third trimester, and 17/18 two weeks postpartum. The authors concluded that the higher dose is appropriate in the third trimester of pregnancy and should be considered during the second trimester and LPV exposure is already back to normal at 2 weeks postpartum. Performing intensive PK is admirable but no HIV-RNA was reported in the abstract. I have several questions about the pharmacokinetic end-points of this study. Why was AUC >52 mg*hr/mL chosen as adequate? What happens to protein binding of LPV during pregnancy and could some of the lower concentrations be secondary to decreased protein binding? How does this recommendation of 533/133 affect the new formulation of Kaletra ? The previous study by Lyons et al. suggested that standard dose of Kaletra is adequate. Possibly the use of TDM and stressing strict adherence is what is needed during pregnancy. I am unconvinced that higher dose of Kaletra is necessary for everyone during pregnancy unless there is some reduction in LPV viral susceptibility at baseline.
Tenofovir nephrotoxicity
Thompson et al. presented data from ESS40006, a salvage study using RTV-enhanced APV with subjects assigned either EFV or TDF based on NNRTI experience (abstract 777). Thus these two groups were different only in the use of EFV or TDF. GFR was followed for 48 weeks. By week 24, GFR decreased 9.85 ml/min in the TDF group which was maintained till week 48 (-11.07 ml/min). The use of EFV did not result in a change in GFR. Predictors of GFR decline were examined using multiple regression analysis, and the only predictor of a decline in GFR was the use of TDF. This study had small number of subjects (n for EFV = 38, TDF = 76) but clearly suggest that TDF affects renal function. There were no dropouts due to renal events, though.
Guest et al followed 222 patients at the Atlanta VA who were on TDF with serum creatinine (calculated creatinine clearance) and serum phosphorus as a measure of the rate of nephrotoxicity (abstract 778). During the first year of treatment 4% of the subjects developed some level of renal insufficiency and 13% had single low serum phosphorus. The composite of serum phosphorus and creatinine clearance abnormality was considered nephrotoxity (how valid is this?) Risk factors for nephrotoxicity were the following: ART naive, amphotericin B use, IV drug user, and ddI as concomitant drug. This was a very drug-experienced population.
Heffelfinger et al. from CDC used the database from CDC's Adult/Adolescent Spectrum of HIV Disease project containing 11,363 HIV-infected subjects receiving care to evaluate the effect of therapy on renal function (abstract 779). Only subjects with normal renal GFR prior to therapy were included. Calculated GFR (Cockcroft-Gault Equation) was used for renal function evaluation. During follow-up, 35.1% experienced mild (GFR 60-89 ml/min), 6.4 % moderate (30-59 ml/min) and 2.6% severe (<30 ml/min) renal dysfunction. Subjects prescribed TDF were more likely to develop renal dysfunction as compared to subjects not on TDF. In a multivariate analysis the odds ratio was 1.6 for mild, 1.5 for moderate renal dysfunction with TDF use, but severe renal dysfunction did not factor out with TDF use. In this large, retrospective, observational study TDF use was associated with renal dysfunction. Other risk factors for renal dysfunction were diabetes mellitus, hypertension, low CD4 count, and low hemoglobin concentration.
Crane et al. examined renal function change in subjects receiving TDF over a 4 year period (abstract 780). 497 patients were identified on TDF and 87 had some level of renal dysfunction (most of which were in the moderate category). In multivariate analysis the use of ddI, age >50 years, and APV use were the associated risk factors with renal dysfunction. Black race was protective. The weakness of this study is that there is no control group not receiving TDF during this time period to assess the actual contribution of TDF to renal dysfunction in these HIV-infected subjects on therapy.
The safety of TDF was evaluated retrospectively in subjects participating in the Viread Early Access Program in the US, Europe, Australia, and Canada (abstract 781). Over 1600 patients had serum creatinine and serum phosphorus in this EAP. The rate of serious renal events was very low: renal failure = 0.3%, Fanconi/tubular disorder = 0.05%, elevated serum creatinine = 0.1%. Elevated serum creatinine resolved 29 days after stopping TDF.
I have the following take from these studies. Renal dysfunction can clearly occur with TDF use but this is very rare and in the general population TDF has proved to be quite safe. However renal function should be routinely monitored after initiation of TDF as part of the overall safety evaluation of all HAART. Mild increases in serum creatinine may not necessarily require cessation of therapy but renal dysfunction is clearly reversible. The use of ddI was identified as an independent risk factor in development of renal dysfunction with TDF, thus TDF and ddI should not be used concomitantly. The adverse drug-drug interaction issue and CD4 lymphocytopenia associated with ddI-TDF concomitant use would further suggest that the use of this drug combination is not prudent.
Relevant to TDF toxicity is the interesting data presented by Kiser et al. (abstract 570) which demonstrated that subjects on Kaletra and TDF had a lower renal clearance of TDF than subjects on TDF but not taking PIs as part of the HAART regimen. The mechanism by which this could occur is unclear. It has been thought that TDF is a substrate of MRP2 exit transporter that is in multiple tissues including the kidney and RTV may be inhibitor of that process thereby decreasing renal secretion of TDF. More recent unpublished data suggest that TDF is a substrate for MRP4 which is not inhibited by RTV. More data need to be generated to resolve these inconsistencies.
Drug-drug interactions
There were numerous drug-drug interaction posters but I will only discuss a few that I believe are important in clinical setting.
With the recent data showing that proton pump inhibitor reduces the exposure to ATV, understanding the effect of acid reducing agents on other protease inhibitor has taken on some importance. Unlike atazanavir, lopinavir has never been shown to have pH-dependent solubility so it was unlikely that drugs that inhibit acid output would decrease the absorption of lopinavir. Nonetheless the study reported by Klein et al. (abstract 578), using Kaletra meltrex formulation, demonstrated that neither omeprazole nor ranitidine affected the pharmacokinetics of lopinavir in HIV-seronegative volunteers. In addition, these investigators confirmed that atazanavir exposure is reduced by both omeprazole and ranitidine. Thus subjects on potent gastric acid reducing agents can safely use Kaletra meltrex as the PI in an antiretroviral combination regimen.
Pujari et al. (abstract 574) studied the interaction between nevirapine and rifampin in HIV-seronegative volunteers in India. A generic version of nevirapine was used in this study. Unfortunately a single dose nevirapine was compared before and after 7 days of rifampin. The authors found that nevirapine exposure was reduced by 79% and they conclude that nevirapine should not be used with rifampin. I disagree with this recommendation because of a single dose nevirapine will not correctly predict the extent of this interaction when nevirapine is brought to steady state. Nevirapine induces its own metabolism thus the concentration of nevirapine is lower at steady state than after a single (non-induced) dose. The effect of rifampin on multiple dose nevirapine pharmacokinetics has been reported and there was only a modest reduction in nevirapine exposure, similar to what has been found with efavirenz. Thus excluding nevirapine use with rifampin is not correct based on other drug-drug interaction data (Ribera et al. JAIDS 28:450, 2001; Robinson et al. abstract 60623, XII World Congress on AIDS, Geneva, Switzerland, 1998).
Scholler et al. (abstract 583) studied the effect of tipranavir/RTV on the pharmacokinetics of TMC-125, the Tibotec NNRTI, in HIV-seronegative volunteers. This study was important because TPV/RTV has numerous drug-drug interaction issues but the package insert indicates that NNRTI, EFV and NVP, pharmacokinetics are not significantly affected by TPV/RTV. However TMC-125 pharmacokinetics was very significantly affected by TPV/RTV. The AUC12hrs was decreased by 76%. On examining the PK curves closely, it appears that most of the effect of TPV/RTV on TMC-125 was on bioavailability as the achieved Cmax was reduced equivalently to AUC. Since TPV is a known inducer of drug transporters, these data suggest that this drug-drug interaction may be on that basis. However I have not seen any data on whether TMC-125 is a substrate for the drug transporters. In a related matter, abstract 565 by Marzolini et al. demonstrated in vitro as well as in rats genetically deficient in mdr1 (Pgp transporter) that EFV is not much of a substrate for these drug transporters. This may be the reason that EFV pharmacokinetics is not altered by TPV/RTV.
Other drug-drug interaction presentations of interest were abstract 585 by Pham et al. demonstrating that Kaletra can be combined with atazanavir with therapeutic concentrations of both lopinavir and atazanavir.
King et al., abstract 586, reported on the ASPIRE II pharmacokinetic data in HIV-seronegative volunteers demonstrating that ATV cannot compete with RTV in enhancing the PK of SQV. However the most interesting data from this study was the demonstration that trough concentration of ATV was at least twice as high when administered as 200 mg b.i.d. as historical data with 400 mg q.d. In addition, sex appears to affect the PK of all three PIs as women achieve higher plasma concentrations when corrected for weight. Abstract 587 examined the pharmacokinetics of once-daily fosamprenavir with ATV 400 mg q.d. in HIV-seronegative volunteers. The data showed that ATV increased APV Cmin almost 3-fold but APV reduced ATV Cmin by -58%. The Cmin of ATV was consistently below 100 ng/mL, which suggests that this drug combination should not be used without RTV enhancement.
Poster 588 generated some controversy as presented by Van Der Lee et al. during the poster discussion session. The investigators planned to examine the effect of Kaletra on the pharmacokinetics of the new statin, Crestor (rosuvastatin) in HIV-infected subjects. The investigators only examined the trough concentration of rosuvastatin and the comparison was to Ctrough rosuvastatin in HIV-seronegative subjects with normal serum lipids. The authors reported a two-fold increase in rosuvastatin Ctrough which seemed minor compared to the RTV interaction with atorvastatin and simvastatin. Many in the audience, however, had concerns about a two-fold increase in Ctrough of rosuvastatin even though the actual control group was not identical to the study population. I found the reaction of the audience concerning since this study never definitively answered the question whether Kaletra affects the pharmacokinetics of rosuvastatin. A study in HIV-seronegative volunteers, where each person serves as his/her control is underway in order to accurately assess the effect of Kaletra on the pharmacokinetics of rosuvastatin. I do not believe that Kaletra will have a significant effect on the PK of rosuvastatin but only the results of the properly designed drug-drug interaction trial will be definitive.
In abstract 575c, Boffito et al described the pharmacokinetic interaction between TMC-114/r and TMC-125 in patients being treated with this drug combination. TMC-114/r reduced TMC-125 exposure by 30% but TMC-114 exposure was unchanged. Frankly I am confused about the human metabolism of TMC-125 as drug-drug interaction studies do not add up. The drug is very lipophilic and undergoes oxidative hydroxylation prior to conjugation. Although the oxidation is thought to be through CYP3A-based on IDV and Kaletra drug interaction studies, but then why doesn't TMC-114/r increase TMC-125 exposure? Hopefully Tibotec will soon examine the ability of a host of transporters to affect the PK of TMC-125.
Sanctuary Site Pharmacokinetics
One aspect of HIV clinical pharmacology with a few key presentation and clinical relevance worthy of a discussion is the importance of antiretroviral drug penetration into sanctuary sites. It has been proposed without much clinical data that the clinical outcome of patients being treated with antiretroviral agents that penetrate sanctuary sites, like central nervous system (CNS) and genital tract, may be better. This has been very difficult to prove definitively but excellent research continues to be performed in the field.
Abstract 74, presented by Letendre et al. from the CHARTER study, examined the correlation between cerebrospinal fluid (CSF) HIV-RNA concentration and being on HAART regimens with good CSF penetration. Penetration scores were assigned to each drug based on a combination of pharmacologic properties, CSF concentrations, and clinical data. Drugs with low penetrations were assigned a score of 0, intermediate penetration a score of 0.5, and high penetration a score of 1. The data collected from 374 subjects indicated that HAART regimens with higher penetration scores are associated with lower CSF HIV-RNA. Since high plasma HIV-RNA and low CD4 count correlated with higher CSF HIV-RNA and higher penetration scores correlated with being on larger number of drugs, a multivariate regression analysis was performed to determine the independence of penetration score on CSF HIV-RNA. The authors reported that higher penetration scores were independently associated with lower CSF viral load (r2 = 0.34, p <0.0001) when adjusted for total number of drugs and plasma viral load. What these data did not have is clinical outcome associated with lower CSF viral load. The most important question that can be evaluated only prospectively is whether there is a difference in the clinical outcome of patients with equivalent plasma viral load on HAART but on drugs with different CSF penetration. Related to this question is abstract 576 by Best et al. demonstrating that the CSF penetration of atazanavir is poor with CSF concentrations on the average 1% of the plasma concentrations. For a drug that is only 86% protein bound in plasma, this CSF concentration is very low and frequently below the viral IC50.
As with the central nervous system, the importance of genital tract penetration of HIV drugs in reducing the viral load in this site is unclear. There is good evidence that lower plasma viral load is associated with decreased heterosexual transmission of HIV (Quinn et al. NEJM 342:921, 2000). However there is no definitive evidence, as of yet, that in HAART-treated subjects drugs that penetrate the genital secretion will result in a greater decrease in genital tract HIV-RNA and therefore reduce sexual transmission of HIV. Dumond et al. (abstract 129) presented data on the concentrations of various antiretroviral drugs in the female genital tract secretion obtained by aspiration. The ratio of genital tract to plasma exposure of these drugs was variable. Nucleosides, which are not highly protein-bound, tend to achieve high ratios while drugs like EFV, which is highly protein-bound, showed a low ratio. The outliers were stavudine and abacavir both of which are not highly protein bound but still had significantly lower genital tract concentrations than what is measured in plasma. Only a prospective study comparing genital HIV-RNA with regimens that have high genital concentrations vs. low genital concentrations will be able to answer the question regarding the relevance of genital penetration for site-specific antiviral efficacy. However to make this issue even more complex understanding of protein binding of these drugs in genital secretions may confound interpretations. In addition, for nucleoside RTIs the rate of phosphorylation may be potentially different at these sites compared to blood. The same research group presented by Vourvahis et al. (abstract 569) demonstrated that tenofovir achieves good genital penetration in both male and female subjects.
Miscellaneous Topics
Owen et al presented an interesting poster that confirms that CYP2B6 expression variation can be measured in tissues other than the liver (abstract 765). The authors looked at the CYP2B6 mRNA expression in peripheral lymphocytes in 20 healthy volunteers and correlated the mRNA to specific single nucleotide polymorphism (SNP), G516T and C1459T. These data are important because it appears from published work by Haas et al. (AIDS 18:2319, 2004) that efavirenz (EFV) pharmacokinetics correlate to the presence of CYP2B6 G516T SNP. CYP2B6 is the main cytochrome P450 isoform that metabolizes EFV. Subjects that are homozygous TT have slow EFV clearance. Interestingly CYP2B6 that has G516T substitution with Gln172His amino acid change express a very functional CYP2B6 in vitro (DMD 31:398, 2003). However these data by Owen et al. demonstrate that the slow EFV clearance may be secondary to decreased expression of the mRNA for the enzyme. Thus there is likely a decreased CYP2B6 content in tissues. Although the authors conclude that EFV's antiviral activity may be affected by the expression of CYP2B6 in lymphocytes resulting in local metabolism, there is no evidence for these data since the investigators provide no measure of actual CYP2B6 content in lymphocytes as compared to liver cells.
Swindells et al. demonstrated that the use of ATV/RTV as maintenance monotherapy is effective in keeping HIV-RNA below level of detection in the majority of subjects in this small exploratory trial and the few subjects that failed therapy did so because of non-adherence as demonstrated by undetectable ATV concentrations without evolution of PI resistance (abstract 108LB). The importance of this study is that simplification of therapy with the use of several RTV-enhanced PIs is possible since the virus has difficulty in developing high level resistance to these drugs. ATV has an advantage over LPV in that the resistance mutation with this PI preserves susceptibility to other PIs but Kaletra has the advantage of having RTV and LPV in a single pill thus selective drug adherence is not possible. Nonetheless perfect adherence to these simplified regimens still rules the day.
CONCLUSIONS
I tried to cover what I considered important pharmacology topics from the 2006 CROI but could not cover everything. I feel that I highlighted the most important and controversial topics. I tried to analyze the data objectively based on my experience and education in Clinical Pharmacology and HIV Medicine. I feel that drug-drug interaction topics continue to take central stage in many of these meetings. The long term safety of antiretroviral drugs remains an important aspect of clinical pharmacology. The poster discussion regarding tenofovir nephrotoxicity was an important aspect of understanding drug safety issues. Complete understanding of pharmacologic issues during antiretroviral combination therapy is the only way we can optimize efficacy and minimize toxicity of these drugs and result in improved clinical outcomes.
|
|
| |
| |
|
|
|
|
|