 |
|
|
| |
Brecanavir: new PI, resistance report from phase II study
|
| |
| |
Reported by Jules Levin
XV Intl HIV Drug Resistance Workshop
June 13-17, 2006
Sitges, Spain
"Resistance-Associated Amino Acid Substitutions and Drug Susceptibility Analysis of Virus from Subjects Entering the Phase II Dose-Ranging Study of a New Protease Inhibitor (PI), Brecanavir, HPR20001 (STRIVE)"
C Craig1, P Yates1, J Flemming2, J Horton3, H Zhao3 on behalf of the STRIVE Study Team 1GlaxoSmithKline, Stevenage, UK; 2GlaxoSmithKline, Greenford, UK; 3GlaxoSmithKline RTP, NC, USA
ABSTRACT
Objective: To analyse baseline genotype/phenotype of brecanavir, a high-affinity binding PI with picomolar potency, in HPR20001 (STRIVE), a Phase II dose-ranging study.
Methods: Baseline genotypes and drug susceptibilities (PhenoSense) from 119/147 subjects were analysed. Correlations between PI mutations, (current IAS.USA defined), and PI susceptibilities were examined. Indinavir was omitted for assay formatting reasons. Recursive partitioning analysis was performed using major PI mutations to predict effects on fold change (FC).
Results: The most prevalent PI mutations were at codons 46 (n=87/119), 90 (n=78/119), 82 (n=74/119), 84 (n=47/119) and 33 (n=47/119). Major PI mutations at positions 30, 48, 50 (V and L) and 88 were underrepresented (n = 5, 6, 5, 2 and 0 respectively). The mean number of major PI mutations/isolate was 3.2
(range: 0 - 7) with mean total PI mutations 11 (range 5-17). Median IC50s for other PIs were significantly above that for brecanavir (median IC50 nM: brecanavir:0.5; amprenavir:230, lopinavir:270, saquinavir:117, ritonavir:1700, nelfinavir:340, atazanavir:74, tipranavir:250).
Median FC for brecanavir was lower than other PIs apart from TPV (median FC: brecanavir:5.9, amprenavir:21, saquinavir:29, nelfinavir:59, ritonavir:162,
lopinavir:82, atazanavir:69, tipranavir:3.4). At clinically derived cut-offs for lopinavir/ritonavir (10-FC), atazanavir/ritonavir (5.2-FC) and tipranavir/ritonavir (4-FC or 2-FC), 83%, 87%, 40% and 65% of isolates were resistant to these PI respectively. For brecanavir, proportions above 10-, 5.2-, 4- and 2-FC were 35%, 55%, 61% and 79% respectively. The mean number of major PI mutations for isolates with FCs ≥ and < cut off were: 3.4/2.2, 3.4/1.9, 3.8/2.8 and 3.5/2.7 for lopinavir, atazanavir and tipranavir (4- and 2-fold) respectively; and 4.0/2.7, 3.8/2.5 and 3.6/2.5 and 3.4/2.3 for brecanavir at the same FCs (10, 5.2, 4 and 2 respectively).
Discussion: Brecanavir shows greater intrinsic antiviral activity than the other PIs tested, and lower FC than all PIs tested except tipranavir/ritonavir, which displays relatively low intrinsic potency and low clinical cut-off. The number of major PI mutations appears to differentiate viruses above and below thresholds. Preliminary analysis of this relatively small dataset suggests that specific mutations associated with increased FC might reflect in part association with higher numbers of mutations. These hypotheses are undergoing clinical
evaluation in ongoing trials.
[This poster presents the analysis from an update of this data set.]
AUTHOR DISCUSSION
The current analysis describes the viruses observed at study entry for the majority of subjects and explored the relationship of genotype to BCV phenotype (drug susceptibility). In addition, exploratory decision tree analysis was performed to try to identify those mutations associated with increased fold change.
The population of viruses had several major and minor mutations present. However, the median IC50 observed with BCV remained sub-nanomolar, which indicates the retention of a very high intrinsic in vitro potency even in the face of multiple PI RAMs. This level of activity was of the order of hundreds to thousands of fold greater than the levels observed with the other PIs.
In terms of fold change, BCV showed the lowest fold change for the PIs apart from TPV, and, like TPV, it showed a relatively small inter-quartile range of activities.
As the virus population was relatively narrowly focused on highly protease-mutated viruses, the decision tree analyses might have been confounded in part by the high numbers of mutations present, making isolation of the effect of any one mutation difficult to assess. In addition, the population showed various imbalances of prevalence of mutations - e.g. >90% with mutations at
codon 10; <10% with mutations at residues 30, 48, 50 and 88.
The analysis suggested that I84V was associated with high fold change observed in this viral population. However, this mutation was found in viruses with fold changes as low as 1.34. Indeed, in a separate survey, I84V present in 10 viruses with varying numbers of other PI RAMs had a mean fold change of 3.8, and 2 viruses showed fold changes <2.5 [Yates, 2005]. Higher levels of fold change were observed with I84V in the presence of I47V, which was indeed associated with higher overall numbers of mutations and this could be a confounding factor. A similar finding of an association between 147V and greater numbers of PI resistance-associated was reported recently [Bethune, 2006].
It should be noted that the analysis examines cross-resistance of BCV to mutations selected by other PIs and the mutations identified do not necessarily predict a resistance pathway for BCV. Indeed, I84V was not selected during sequential in vitro passages of virus in the presence of BCV [Yates, 2006].
Further detailed analyses to determine relatedness between particular patterns of resistance and fold change are ongoing.
Author Conclusions
The population recruited into the BCV dose-ranging clinical study, HPR20001 (STRIVE) shows a high degree of prior selection of PI resistance-associated mutations and high levels of resistance to all currently approved PIs except TPV.
The PI resistance-associated mutation, I84V, was observed in association with high fold change, although there were overall slightly more PI resistance-associated mutations in the viruses with I84V than in the wild-type I84-containing viruses, particularly when I47V was also present.
Despite the multi-PI resistance mutations, the majority of viruses (68%) from subjects at Day 1 showed sub-nanomolar IC50 BCV activity.
Introduction
In order to build on the initial success of combination antiretroviral therapies (ART) against HIV-1 infection, it is important to continue to develop drugs to address the needs of highly ARTexperienced individuals, whose viral infection may be poorly controlled due to reduced drug susceptibility. Over time, the number of these individuals and the extent of their drug resistance are
increasing.
Two main strategies are currently being employed to address these issues. First, drugs are being developed against new antiretroviral targets, where resistance has not yet been selected. Secondly, new drugs are being developed against the fully validated targets such that the new drugs suffer relatively little cross-resistance to viruses that have several resistance-associated mutations from
previous selection during treatment with earlier drugs of the same class is observed. Brecanavir (BCV, GW640385) is a product of the second of these approaches. Brecanavir exhibits a superior binding affinity compared with previous protease inhibitors (PIs), with a Ki 15 x 10-15M [Hazen, 2003]. In addition, BCV shows a very high intrinsic potency in vitro (geometric mean IC50:
clinical isolates in PBMC 0.03 nM - n = 26; HXB2 strain in MT4-MTT assay: 0.66 nM - n = 27, data on file).
The objective of the current study was to further characterise genotype and phenotype (drug susceptibility) relationships for BCV in the context of other PIs, assessed in parallel with the same genotypes using the day 1 genotype/phenotype data from the clinical trial HPR20001 (STRIVE).
HPR20001 is an on-going Phase IIB, randomized, multicenter, parallel-group study to evaluate the safety, pharmacokinetics and antiviral effect of four blinded dosing regimens of BCV/ritonavir (/r) therapy compared to open-label current PI/r therapy in HIV-1 infected, PI experienced adults.
Methods
A total of 147 subjects were enrolled into HPR20001 and to date, Day 1 genotype and phenotype have been attempted for 140 subjects. This has been supplemented with 5 screening data sets (total n=145). Analysis of the remaining two samples is on-going. The enrolment criteria required the presence of two or more multi-PI resistance mutations: L10I/F/V/R, V32I, M46I/L, I54V/L/M,
V82A/F/T/S, I84V/A/C, L90M [Johnson, 2004].
Sequence analysis was performed at Monogram Biosciences, Inc., South San Francisco, CA., by a thermocycling method using fluorescent dye labelled dideoxynucleotide chain terminator chemistry. Resistance-associated mutations were classified based on the IAS USA resistance table [Johnson,
2005]. All assays were performed by scientists at Monogram Biosciences, Inc., South San Francisco, CA [Petropoulos, 2000]. The mean percent inhibition for each drug concentration was determined and used to calculate the IC50. The fold change (FC) in drug susceptibility was determined by comparing the IC50 for the subject virus to the IC50 for the drug-sensitive reference virus containing the PRO and RT sequences of the NL4-3 strain of HIV-1. For the purposes of analysis, the data were censored at the maximum concentration tested when the IC50 or FC exceeded this concentration.
Decision tree analysis was carried out using a recursive partitioning method to examine the pathways of cross-resistance that affect drugs using HelixTree software, where the drug phenotype resistance was dependent variables and the mutations codons were predictors in the model.
RESULTS
Of a total of 147 enrolled subjects, Day 1 genotypes were available for 134, and genotypes obtained at the time of screening were available for five subjects. Genotypes were not obtained for 6 subjects and two were in progress and were not included in the analysis. Overall, the mean number of major PI resistance-associated mutations (RAMs) was 3.2 (range 0-7) and of total PI RAMs 11.2 (range 4-17).
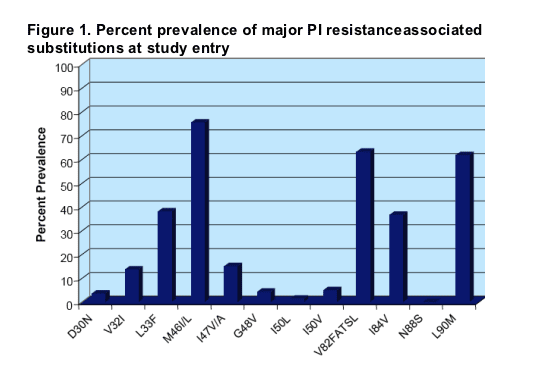
PI resistance-associated substitution
The most prevalent major substitutions included M46I/L (76%), V82FASTL (63%) and L90M (62%). It is also notable that >30% of viruses also included I84V and/or L33F. Mutations at codons 30, 48, 50 and 88 were present at low prevalence.
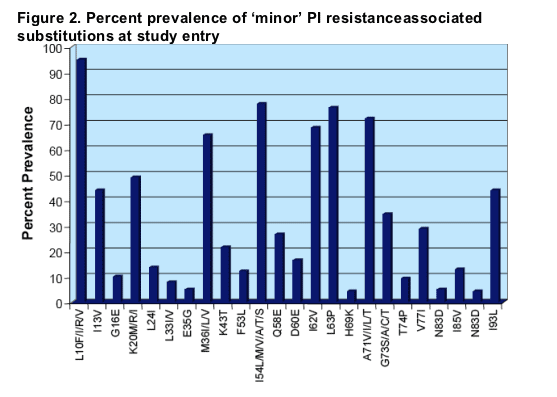
PI resistance-associated substitution
A high prevalence of minor mutations was also observed (e.g. >90% substitutions at residue 10 and >70% at residues 54 and 71). These findings are characteristic of a very highly PI-experienced population.
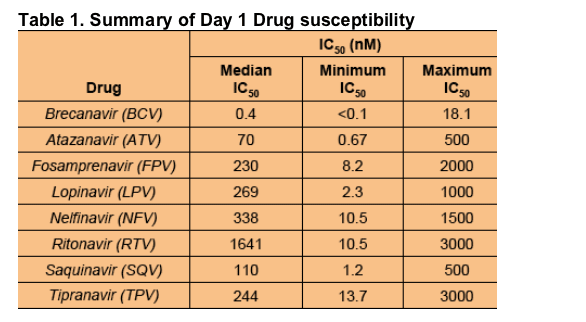
The median IC50 for BCV remained sub-nanomolar for this highly mutated population of viruses, while other PIs, showed IC50s in the range 70 - 1641 nM (175 - 4100 fold higher) with TPV 610-fold higher. Indeed, four viruses were found with BCV IC50 <0.1 nM. The maximum values indicate the highest concentration tested, and for all other PIs, the IC50 was higher than those
observed with BCV.
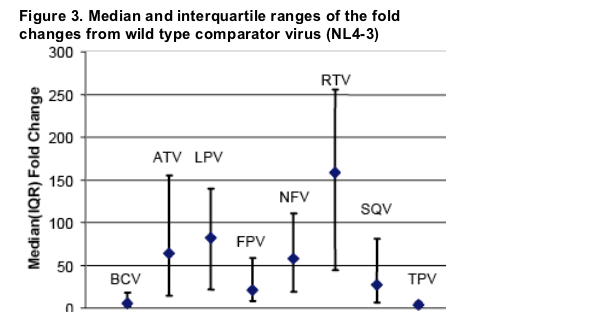
Both BCV and TPV show a low overall level of fold change relative to the other PIs. In addition, these two compounds show a limited range of fold change around the median, which also supports the suggestion that these compounds more readily retain their intrinsic in vitro potency levels (wild type IC50: BCV: 0.1 nM; TPV: 76 nM; fold change: BCV: 5.7; TPV: 3.2) even in the face of multiple PI resistance-associated mutations.
The next least affected PIs were FPV and SQV, although the inter-quartile range for FPV is slightly less than for SQV.
Figure 4. Decision tree analysis
In order to identify specific mutations contributing to resistance to BCV, a decision tree analysis was performed using the full protease sequences from all 138 viruses with drug susceptibility data.
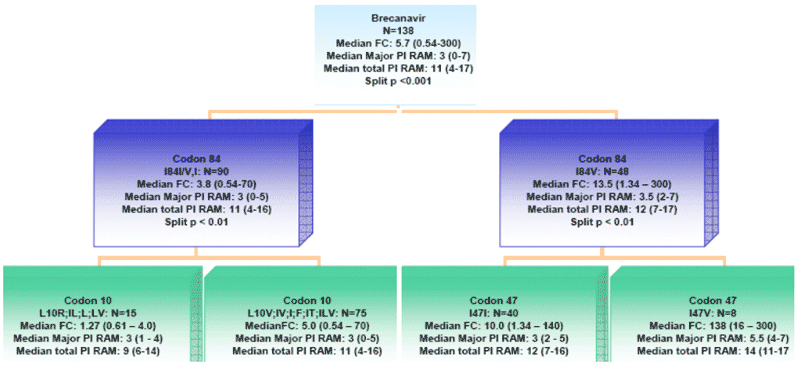
A preliminary decision tree analysis using all 99 codons in protease resulted in determination of three splits on two levels - one around codon 84, another at codon 10 (with mostly wild type I84) and codon 47 (with mutant I84V). The first split was statistically significant, even after Bonferroni adjustment. However, the splits for codon 10 and codon 47 were statistically significant only before Bonferroni adjustment.
A similar pattern was observed when only the major and minor PI RAMs were used. However, when only major mutations were used, the split at codon 10 (a minor mutation) gave way to a split at codon 47 (I versus A, V, L), but this was not statistically significant after Bonferroni adjustment.
References
Bethune et al., 4th EHDRW March 29-13, 2006 Monte Carlo, Monaco, Abstr # 51.
Florance et al., XIII IHDRW June 8-12, 2004, Tenerife Sur-Costa Adeje, Canary Islands, Spain, Abstr # 11.
Hazen et al., 2nd IAS Conf on HIV pathogen and Treat, Paris, France, Jul 13-16, 2003 Abstr # 541.
Johnson et al., (2004) Update of the Drug Resistance Mutations in HIV-1: 2004. Top HIV Med. 12:119-124.
Johnson et al., (2005) Update of the Drug Resistance Mutations in HIV-1: Fall 2005, Top HIV Med. 13:125-131.
Petropoulos et al., (2000) Antimicrob Agent Chemother. 44:920-8.
Yates et al., 10th EACS Conference, Dublin, Ireland, 17-20 Nov, 2005 Abstr # PE3.3/3.
Yates et al., (2006) Antimicrob Agent Chemother 50:1092-1095.
|
| |
|
|
|
|
|