 |
|
|
| |
Pfizer Tropism Assay: Evaluation of an Ultra-deep Sequencing Method to Identify Minority Sequence Variants in the HIV-1 env Gene from Clinical Samples
|
| |
| |
Reported by Jules Levin
CROI, Feb 2007, Los Angeles
Marilyn Lewis*1, I James1, M Braverman2, B Desany2, T Jarvie2, M Penny1, R Harrigan3, M Youle4, R Hernandez1, and E Van Der Ryst1
1Pfizer Global R&D, Sandwich, UK; 2454 Life Sci, Branford, CT, US; 3BC Ctr for Excellence in HIV/AIDS, Vancouver, Canada; and 4Royal Free Hosp, London, UK
Background: The aim of this study was to identify minority HIV-1 env sequences in a pre-treatment sample of a patient identified as having only R5 HIV at screening in whom CXCR4-using virus was detected following 10 days of monotherapy with maraviroc (MVC). The 454-technology simultaneously sequences hundreds of thousands of clones within a single sample using an emulsion-based method to amplify and immobilize DNA fragments spanning the gene of interest. This method should therefore detect individual clones present in a heterogeneous virus pool at a frequency of <1%.
Methods: Amplicons of the full-length env gene (2.5 kb) from 2 plasma samples (days 1 and 11) were randomly fragmented and sequenced using a 200-bp average read length protocol to a depth of >100,000 reads. From the 2 samples the 454-reads were mapped against the HIV-1 sequence database from Los Alamos National Laboratory (LANL). Using the closest match from the database, sequencing reads were incorporated into the existing HIV-1 multiple sequence alignment from LANL. Sequences were assigned a CXCR4-using genotype according to the 11/25 charge rule.
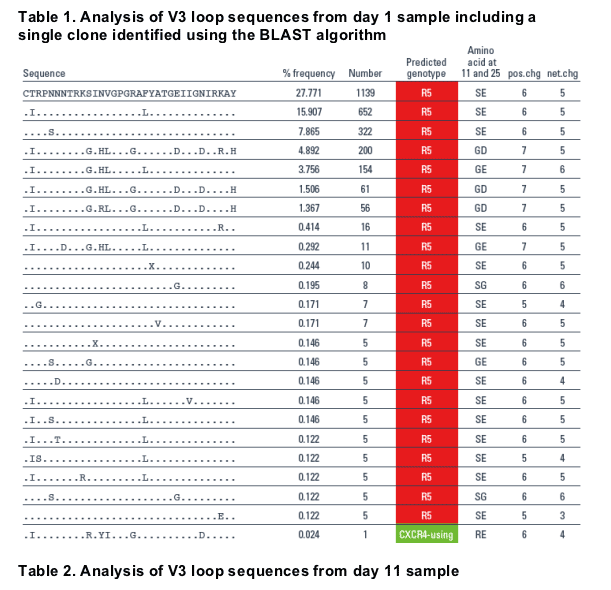
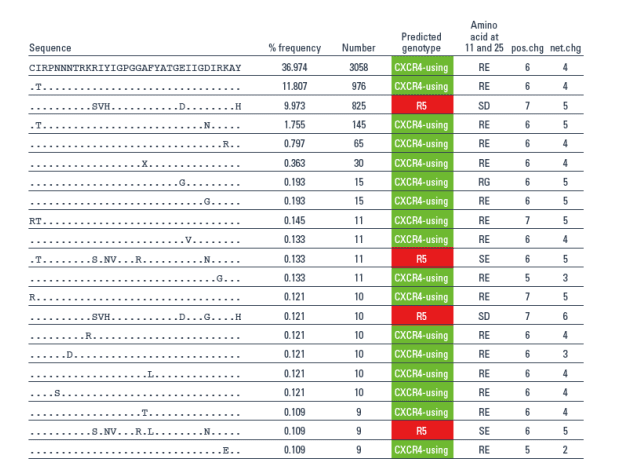
Results: A total of 104,628 and 191,637 reads were obtained for the days 1 and 11 samples, respectively, and compared to the LANL env alignment. In this fashion, >99% of the reads could be mapped. The heterogeneity of env made it difficult to reconstruct full-length quasispecies, so we focused on reads that fully spanned the V3 region. This resulted in 4104 and 8275 V3 spanning reads for the days 1 and 11 samples, respectively. The day 11 sample contained 20 unique V3 sequences at a frequency of >0.1%, with 90% of these having a CXCR4-using genotype. In contrast, no CXCR4-using sequence was identified amongst the 23 unique V3 sequences from day 1, present at >0.1% frequency. By using the BLAST algorithm and a typical CXCR4-using sequence from day 11, a single sequence with a CXCR4-using genotype was identified in the day 1 sample (0.02%).
Conclusions: Ultra-deep sequencing provided good-quality sequences and detected minority HIV-1 env sequences in a clinical sample. A CXCR4-using genotypic sequence was detected from the day 1 sample but only following secondary analysis. These results concur with the phenotypic tropism assessment of these samples.
Introduction and Objectives
Maraviroc (MVC) is a novel antagonist of the CCR5 co-receptor, with potent antiviral activity in vitro and in vivo against CCR5-tropic (R5) HIV-1 strains, including virus resistant to currently available antiretrovirals (ARVs).1,2
In two Phase 2 dosing studies, a total of 64 patients pre-screened for the absence of CXCR4-using virus received MVC monotherapy for 10 days.2
Dual-tropic virus was detected post-treatment in a previously treatment-naive patient (Patient A) who had responded clinically to MVC monotherapy (reduction in viral load of 0.71 log10 copies/mL).3
The aim of this study was to evaluate whether an ultra-deep sequencing method4 has the capability to detect individual clones present in a heterogeneous virus pool at a frequency of < 1%, by identifying minority HIV-1 env sequences in pre- and posttreatment samples from this patient.
Methods
Samples were prepared at the BC Centre for Excellence in HIV (Vancouver, BC,
Canada), where RNA extraction and amplification of the gp160 region took place.
Amplicons of the full-length env gene (2.5 kb) from two plasma samples (days 1 and 11) were randomly fragmented and sequenced using a 200 base pair (bp) average read length protocol to a depth of greater than 100,000 reads.
-- The Genome Sequencer 20� System (454 Life Sciences, Branford, CT, USA) is an ultra-high-throughput automated DNA sequencing system capable of resolving hundreds of thousands of clones within a single sample using an emulsion-based method to amplify and immobilise DNA fragments spanning the gene of interest.4 An overview of the 454 sequencing system is shown in Figure 1.
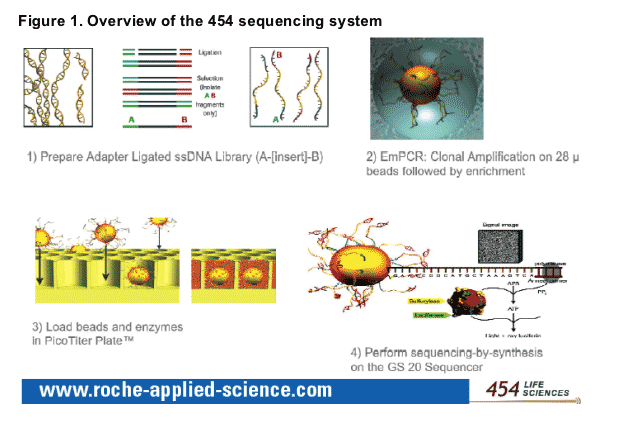
Reads from the two samples were mapped against the HIV-1 sequence database from Los Alamos National Laboratory (LANL). Using the closest match from the database, sequencing reads were incorporated into the existing HIV-1 multiple sequence alignment from LANL.
The section of the alignment corresponding to the V3 region was extracted and any individual sequence species present at less than 0.1% were removed from the analysis in order to remove noise contributed by sequencing error.
Redundant sequences were then collapsed into one sequence, for convenience in subsequent steps.
The resulting sequences after this filtering and collapsing were re-aligned with ClustalW5, visually inspected and, where appropriate, further hand-corrected for sequencing errors due to low quality regions (e.g. homopolymeric regions where over- or under-calling resulted in a frameshift).
The resulting sequences were translated in the appropriate frame, collapsed into
non-redundant amino-acid sequences, and aligned for a final time with ClustalW.
V3 loops sequences were assigned a CXCR4-using genotype on the basis of the
position-specific scoring matrices (PSSM;
http://ubik.microbiol.washington.edu/computing/pssm/)6 and the presence of basic amino acid residues at positions 11 and/or 25.7,8
RESULTS
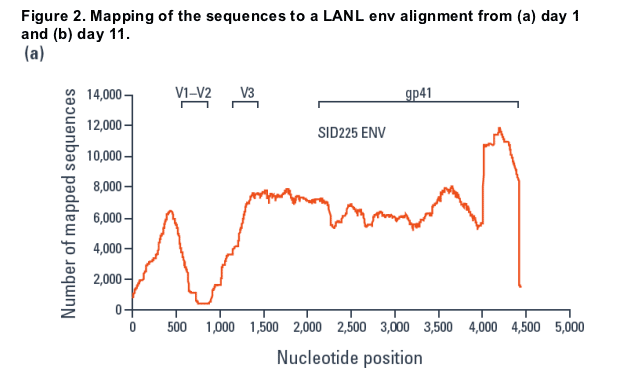
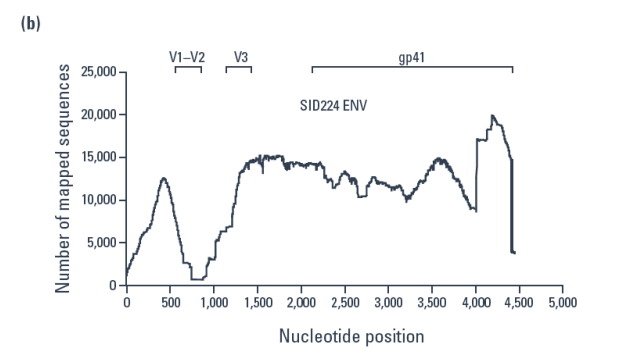
A total of 104,628 and 191,637 reads were obtained for the day 1 and 11 samples, respectively, and compared to an env alignment from the LANL database (Figure 2).
- Using the closest match from the database, sequences were mapped using alignments as small as 16 bp, resulting in over 99% of the reads being mapped.
- A dip in the mapping over the V1/V2 region and an increase at the 3' end was seen.
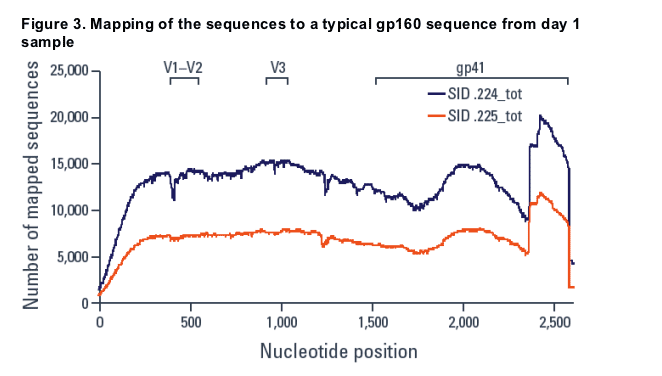
Mapping day 1 and day 11 sequences to a typical full length gp160 sequence from the day 1 sample (Figure 3), with parameters requiring any individual alignment to be over at least 80% of the read length and at least 80% similar to the reference sequence, resulted in approximately 80% of sequences being mapped.
- This eliminated the dip in the coverage of the V1/V2 region but the increase at the 3' end remained.
Since the heterogeneity of env made it difficult to reconstruct full-length quasispecies, focusing on reads that fully spanned the V3 region resulted in 4,104 and 8,275 V3-spanning reads for the day 1 and 11 samples, respectively.
- The day 11 sample contained 20 unique V3 sequences at a frequency of > 0.1%, with 90% of these having a CXCR4-using genotype (Table 2).
- In contrast, no CXCR4-using sequence was identified amongst the 23 unique V3 sequences from day 1, present at > 0.1% frequency.
A secondary analysis was carried out using the BLAST algorithm and a typical
CXCR4-using sequence from day 11 and led to the identification of a single sequence with a CXCR4-using genotype in the day 1 sample (0.02%, Table 1).
CONCLUSION
Ultra-deep sequencing provided good quality sequences across the entire gp160
region and detected minority HIV-1 env sequences in a clinical sample.
The genotypic sequences derived from the two samples were consistent with the phenotypic tropism assignment at each timepoint.
Detailed sequence analysis of this pre-treatment sample using ultra-deep
sequencing did not predict emergence of the dual-tropic phenotype.
References
1. Dorr P et al. Antimicrob Agents Chemother 2005;49:4721-32.
2. Fatkenheuer G et al. Nat Med 2005;11:1170-2.
3. Westby M et al. J Virol 2006;80:4909-20.
4. Margulies M et al. Nature 2005;437:376-80.
5. Chenna R et al. Nucleic Acids Res 2003;31:3497-500.
6. Jensen MA et al. J Virol 2003;77:13376-88.
7. De Jong N et al. J Virol 1992;66:6777-80.
8. Fouchier RAM et al. J Clin Microbiol 1995;33:906-11.
|
| |
|
|
|
|
|