 |
|
|
| |
Two-Year Follow-up of Treatment-Experienced Patients on Vicriviroc (VCV)
|
| |
| |
Reported by Jules Levin
ICAAC Sept 17-20, 2007, Chicago
As I said in the previous email this was CCR5 day at ICAAC. In a sense this meeting was a 'coming out' party for CCR5, a bit of an exaggeration. But we saw 48 week Maraviroc safety/efficacy data and Vicrriviroc 3-year followup showing no safety surprises. In addition to the Incyte CCR5 and PRO 140 presented here and in Sydney. The chicken was good at the Monogram reception tonight, but the salmon was not very good last night at the Gilead symposium. Wednesday is hepatitis day at ICAAC but I did not see much of interest in the abstract book. Tit seems that HCV research in HIV likes to repeat all the studies conducted long ago and repeatredly in HCV monoinfection. For example, there is a poster here tomorrow showing insulin resistance reduces response to pegf+RBV. Well, that was well established years ago in HCV monoinfection with a considerably large body of research.
R. Gulick1, D. Haas2, A.C. Collier3, J. Lennox4, C. Parker5, W. Greaves5
1Cornell Univ., New York, NY; 2Vanderbilt Univ., Nashville, TN; 3Univ. Washington, Seattle, WA; 4Emory Univ., Atlanta, GA;
5Schering-Plough Research Institute, Kenilworth, NJ
AUTHOR CONCLUSIONS
The results presented here demonstrate the 3-year durability of vicriviroc-containing optimized regimens in heavily treatment-experienced, HIV-positive patients with prior antiretroviral failure.
--These data are the first to demonstrate the sustained effects of a CCR5 antagonist-based regimen through >3 years of therapy.
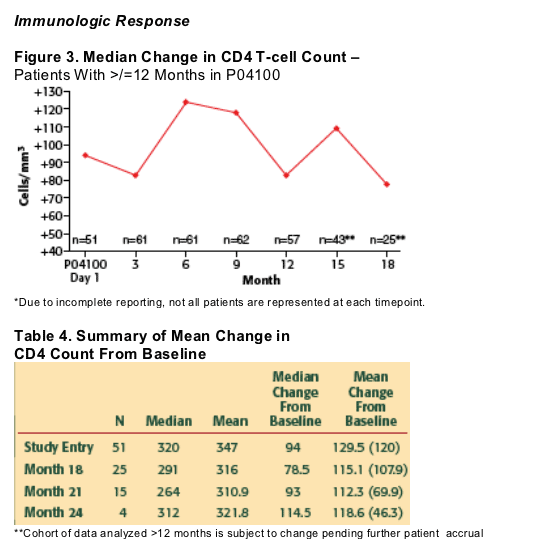
HIV suppression and CD4 T-cell increases were maintained, and opportunistic infections did not occur.
The observed adverse events were not unexpected in this population. There were no reported treatment-related hepatic or cardiovascular toxicities.
ABSTRACT
Background: VCV demonstrated durable antiretroviral activity and CD4 response in treatment-experienced (TE) subjects at 48 weeks (Gulick, IAS 2007 abstract 1623). We present 2-year data for TE patients who received VCV and participated in a roll-over study.
Methods: HIV-1 infected subjects successfully completing the 48-week phase of
ACTG 5211 could enter this open-label multicenter study and receive VCV 15 mg
in addition to previously optimized background therapy (OBT) that included a
ritonavir-boosted protease inhibitor. Additionally, subjects with a tropism shift to
R5/X4 virus with HIV-RNA >/=0.5 log10 below baseline and no decrease in CD4
count could enroll. We assessed change from baseline in HIV-RNA and CD4 in
subjects who had >/=12 months follow-up, as well as new opportunistic infections
(OIs); malignancies; seizures; and hepatotoxicity.
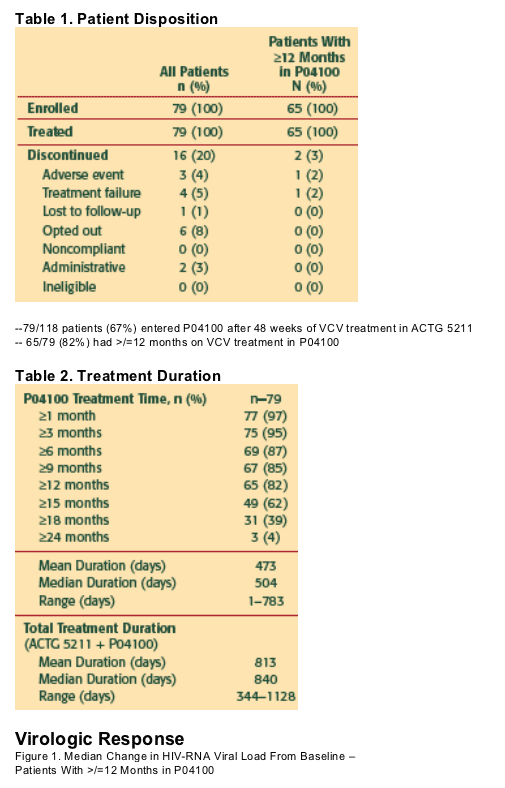
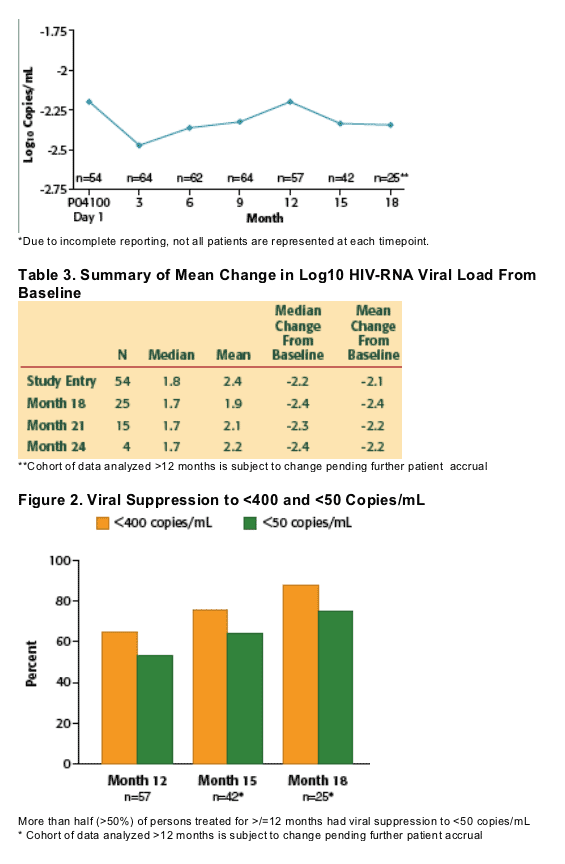
Results: Of 79 subjects entering the study, 54 had at least 12 months exposure to VCV. Data were available for 39 subjects (data pending for 15 ongoing subjects): mean treatment duration (from first dose in ACTG 5211 to last dose in the rollover protocol) was 103 weeks (range 49-145). Median change in HIV-RNA from baseline (ACTG 5211) was -2.2 log10; median change in CD4 count was +84 cells/mm3; 23/39 subjects (60%) had HIV-RNA <50 copies/mL and 28/39 (72%) had <400 copies/mL. 12 subjects discontinued: 2 due to adverse events, 6 for patient choice, 2 for administrative reasons, and 2 for virologic failure. 6 tropism shifts occurred during the study: 2 to X4 and 4 to R5/X4. Other than pulmonary TB (n=1), no OIs, VCV-related hepatotoxicity, cardiovascular events, seizures, or new lymphomas (since February 2006) were reported.
Conclusions: VCV, 15 mg qd plus OBT, was generally well tolerated and provided potent and durable antiretroviral activity. These results represent the longest follow-up data available for a CCR5-containing regimen to date.
BACKGROUND
--Vicriviroc (VCV) is a novel extracellular inhibitor of HIV infection designed to block entry of infectious virions into uninfected CD4 cells via antagonism of the CCR5 co-receptor (CCR5 antagonist).
--VCV plasma concentrations are increased 2-6 fold by CYP3A inhibitors,
including ritonavir.
--VCV plasma half-life >24 hours allows for once-daily dosing.1
--VCV is in Phase 3 development for the management of treatment-experienced
HIV-1 infected patients.
ACTG 5211 was a double-blind, randomized Phase 2 study of vicriviroc in HIV-positive, treatment-experienced patients experiencing virologic failure while receiving a ritonavir-containing regimen.2
- At baseline, patients had HIV-RNA levels >5000 copies/mL and R5- tropic HIV. Patients received their previous regimen plus 5, 10, or 15 mg VCV or placebo for the first 14 days, after which the background antiretroviral combination was altered to an optimized ritonavir-containing regimen. The 5 mg VCV trial arm was discontinued early.
- At 48 weeks, the proportion of patients with virologic failure (<1 log decline in HIV-RNA after >16 weeks) was 86% in the placebo arm, 27% in the 10 mg VCV arm and 33% in the 15 mg VCV arm.3
- Study treatment discontinuations by week 48: 82% (placebo), 30% (VCV 10 mg), 37% (VCV 15 mg).
- Malignancies occurred in 8 patients taking VCV and 2 patients in the placebo arm.3 An independent SMC concluded that a causal relationship with VCV could not be determined.
- Of the 26 patients on VCV who experienced virologic failure, 9 (35%) developed HIV tropism shifts. Their viral populations became CCR5/CXCR4 dual or mixed tropic or solely CXCR4-using.
STUDY OBJECTIVES
To provide subjects previously enrolled in the Phase 2 ACTG 5211 protocol with continued vicriviroc for use in combination with ritonavir-containing background antiretroviral therapy.
To assess the long-term durability, safety, and tolerability of vicriviroc combination therapy in treatment-experienced patients.
METHODS
Trial P04100 is an open label, multicenter, treatment protocol for HIV-infected subjects initially randomized to treatment regimens under ACTG Protocol A5211.
Population: HIV-positive patients who successfully completed 48 weeks in ACTG 5211 (or screened for ACTG 5211 but were unable to enroll due to early protocol closure)
- Eligible patients had either R5-tropic HIV or HIV that in the course of ACTG 5211 underwent a coreceptor tropism shift to R5/X4 virus (dual or mixed tropism).
For patients with a coreceptor tropism shift, HIV-RNA had to be >0.5 log below their ACTG 5211 baseline with no decrease in CD4 count.
--Study participants must have been taking a ritonavir-containing antiretroviral regimen (with no drugs that induce cytochrome P450 3A4).
--Study participants received VCV 15 mg once daily with the optimized background therapy (OBT) prescribed in ACTG 5211.
--Patients were discontinued from the study after confirmed virologic failure (two measurements of HIV-RNA <0.5 log below ACTG 5211 baseline) or immunologic decline (CD4 T-cell count below the ACTG 5211 baseline).
Patients with dual/mixed HIV tropism were discontinued from the study after virologic failure or decrease in CD4 T-cell count to below their ACTG 5211 baseline.
--Monitoring was done at Days 1, 14, 28, and Month 2 and every 2 months thereafter.
--Monitored parameters included CD4/CD8 count, HIV-RNA, vicriviroc susceptibility, serious adverse events, new infections, AIDS-defining conditions, and changes in coreceptor tropism of patient HIV isolates.
--On-treatment analysis of virologic and immunologic response and safety was performed.
RESULTS
16 (20%) patients discontinued P04100 early for any reason
Viral Coreceptor Tropism
6/79 patients entering 4100 with R5 virus experienced a change in coreceptor use during the study
-- 4 patients had dual-tropic (D/M) virus, of which 1 remains suppressed and 1 experienced virologic failure.
-- 1 patient had CXCR4 tropic virus and is continuing on study.
-- 1 patient had D/M virus with a change in coreceptor use to CXCR4 tropic virus and experienced virologic failure.

4 patients experienced virologic failure during the course of this study (two occurred at the time of a change in tropism).
-- 3/4 patients with virologic failure and for whom resistance testing was available, had no evidence of resistance to vicriviroc as determined by percent maximal suppression.
Adverse Events
68/79 (86%) patients experienced treatment-emergent adverse events.
The most common adverse events were nausea: n=17 (22%), fatigue: n=16 (20%), pain in extremities: n=12 (15%), headache: n=12 (13%), cough: n=10 (13%), and diarrhea: n=10 (13%).
The majority of events were Grade 1-2 (mild to moderate in severity); fatigue (3%) was the only Grade 3 adverse event reported in greater 1% of patients.
During the 2 years of this study, no systemic malignancies were reported. Two skin cancers were observed and determined by the investigator as unlikely to be related to vicriviroc. Both patients were caucasian, male, and greater than 48 years of age.
No AIDS-related opportunistic infections, hepatic or cardiac toxicities occurred.
References
1. Sansone A, Keung A, Tetteh E, et al. Pharmacokinetics of vicriviroc are not affected in combination with five different protease inhibitors boosted by ritonavir [abstract 582]. In: Program and abstracts of the 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO, Feb. 5-8, 2006. Alexandria, VA: Foundation for Retrovirology and Human Health, 2006:105.
2. Gulick RM, Zu S, Flexner C, et al. Phase 2 Study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS Clinical Trials Group 5211. Journal of Infectious Diseases. 2007;196:304-12.
3. Gulick R, Zu S, Flexner C, et al. ACTG 5211: Phase 2 Study of the safety and efficacy of vicriviroc (VCV) in HIV+ treatment-experienced subjects: 48-week results. Program and abstracts of the 4th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; July 22-25, 2007; Sydney, Australia. Abstract TUAB102.
|
| |
|
|
|
|
|