 |
|
|
| |
Safety and tolerability of KP-1461 in phase 1, dose-ranging study in highly ART experienced HIV infected persons
|
| |
| |
Reported by Jules Levin
IDSA Oct 2007
P. G. Clay1, D. E. Sweet2, O. O. Osiyemi3, E. Godofsky4, R. Redfield5, S. E. Smith6, R. Campo7, J. H. Shrank8, J. Parkins9, J. Reno9, S. Becker9
1Kansas City University of Medicine and Biosciences, Kansas City, MO, 2University of Kansas School of Medicine, Wichita, KS, 3Triple O Medical Services, West Palm Beach, FL, 4Bach and Godofsky, MD, PC, Sarasota, FL, 5University of Maryland, Baltimore, MD, 6New Jersey Medical School, Newark, NJ, 7University of Miami, Miami, FL, 8Greenville Hospital System, Greenville, SC, 9Koronis Pharmaceuticals, Inc., Redmond, WA.
SUMMARY
KP-1461 is a novel antiretroviral agent that works as a selective viral mutagen through a process called Viral Decay Acceleration. Unlike conventional ART, KP-1461 demonstrates irreversible viral extinction in vitro. This phase I-b study shows that treatment with KP-1461 is generally safe and well tolerated when
administered over 14 days at doses of 400, 800, and 1600 mg every 12 hours to HIV-infected subjects. Whether the notable effect seen in the in vitro studies is replicated in vivo is the subject of an ongoing phase 2 study.
Background
KP-1461 is a novel, first-in-class, therapeutic agent for the treatment of human immunodeficiency virus (HIV) infection. KP-1461 is an oral prodrug of KP-1212, which in vitro irreversibly extinguishes HIV through the process of Viral Decay Acceleration (Harris).
KP-1461 is a deoxycytidine analog that acts as a selective viral mutagen. As a result of its flexible structure, KP-1461 induces base pairing errors. This accumulation of errors leads to a progressive reduction of viral fitness and eventual error catastrophe and population collapse. The naturally high error rate in the incorporation of bases during HIV transcription and the information dense genome allow the virus to escape immune system responses and to develop resistance to antiviral drugs. As a result, HIV exists as a quasispecies containing related variants with differing degrees of fitness, virulence and pathogenicity. The high inherent error rate, lack of RT proofreading capability, and no known ability to repair a DNA-RNA heteroduplex make HIV and other viruses particularly vulnerable to additional errors that could adversely affect viral population survival (Overbaugh; Eigen; Anderson).
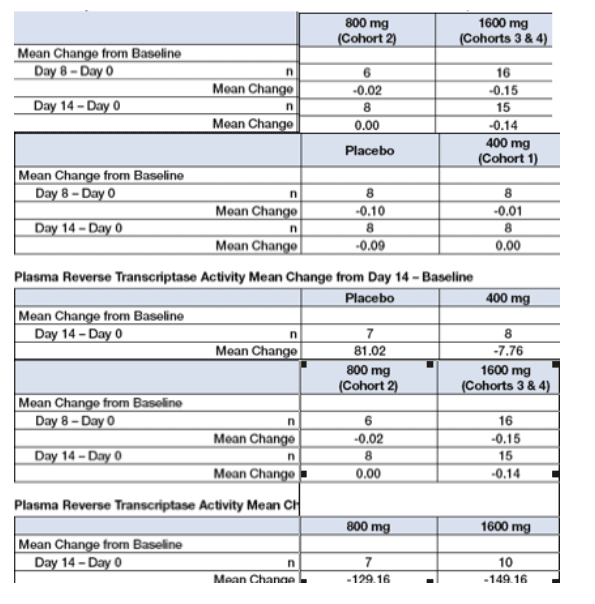
KP-1461 contains an unmodified sugar, allowing continual chain elongation by RT, with a modified base that appears ambiguous to the complementary base. Through a tautomeric process, KP-1461 can pair with either guanosine or adenosine, thus creating G-to-A and C-to-T errors (Loeb). Base pairing errors are incorporated randomly throughout the viral genome and persist through subsequent replication cycles. Based on in vitro data where HIV was ablated, modeling suggests that at least 8 to 12 weeks of treatment may be required before a significant antiviral effect in humans is noted.
Consequent to its mechanism of action, KP-1461 does not appear to exert the same type of selective pressure on the virus as conventional antiretrovirals. This may reduce, or even preclude, the development of KP-1461-resistant variants.
The ability to irreversibly extinguish virus in vitro, a feature that distinguishes KP-1461 from all approved antiretroviral agents, and extensive pre-clinical evaluation conditioned the conduct of the phase I-b study performed in HIV-infected individuals reported here.
Additional Findings
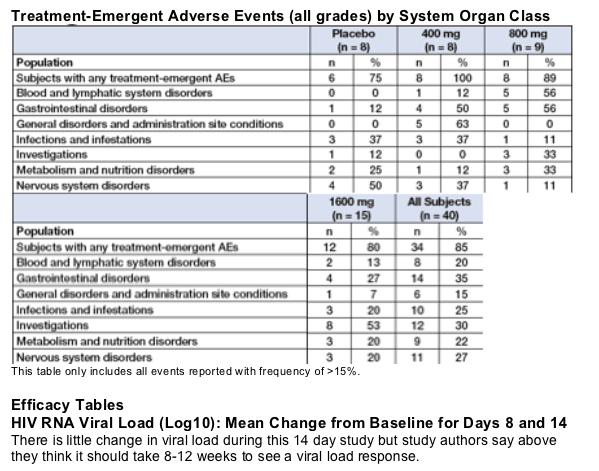
Combining all subjects treated with KP-1461 to evaluate the system organ classes with the highest incidence of events, the most frequently reported adverse events were: Gastrointestinal Disorders (13 subjects, 41%), Investigations (11 subjects, 34%), Blood and Lymphatic Disorders (8 subjects, 25%), Nervous System Disorders (7 subjects, 22%), Infections and Infestations (7 subjects, 22%), and Metabolism and Nutrition Disorders (7 subjects, 22%).
When individual adverse events were evaluated, headache, nausea, neutropenia and fatigue (or increased fatigue) were the most frequent, with 5 events. Other events were thrombocytopenia and diarrhea, each with 4 events, and dizziness, with 3 reported events for the total KP-1461 group.
Discussion
Safety & Tolerability:
These data demonstrate that KP-1461 is generally safe and well tolerated when administered at multiple doses, thus warranting phase 2 efficacy studies.
--Most adverse events were mild to moderate in intensity
--There were no dose-dependency findings to AEs
-- Gradable values for laboratory abnormalities were generally mild to moderate and showed no dose-dependency
--Further evaluation of hematologic findings in longer-term studies is warranted
--No clinically significant changes noted in serial ECG
A total of 3 subjects experienced SAEs. None were attributed to KP-1461
- Thrombocytopenia
- Muscle spasm
- Catheter-related infection
Efficacy:
Despite the expectation that a longer period of dosing would be required, trends are suggestive of antiviral effect by HIV RNA and RT assay at higher doses. (Formal efficacy analysis was not planned as part of this assessment. Descriptive data only is provided.)
Pharmacokinetics:
Findings are supportive of pursuing twice daily dosing. (Full PK data will be presented at European AIDS Conference, Madrid, October 2007.)
Purpose
The purposes of study KP-1461-102 are to determine the safety, tolerability, and pharmacokinetic activity of multiple oral doses of KP-1461 in HIV+ men and women, and to assess any effect of KP-1461 on plasma HIV RNA.
Objectives
To assess the safety and tolerability of oral KP-1461 administered every 12 hours for 14 days (28 doses) to cohorts of HIV+ subjects
To determine the PK profiles of KP-1461 (the inactive prodrug) and KP-1212 (the active drug)
To assess the effects of twice-daily KP-1461 over 14 days on plasma HIV RNA copy number and HIV Reverse Transcriptase activity
STUDY DESIGN
Methods
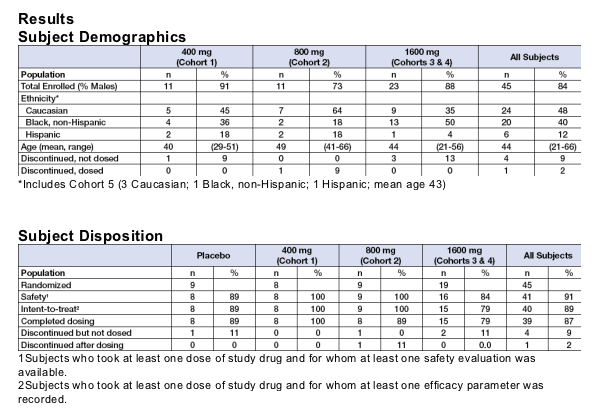
This was a phase 1-b multi-site, randomized, double-blind, placebo-controlled dose-escalation study. Study population included:
• Men and women 18 to 60 years of age
• CD4+ cell counts >100cells/mm3
• HIV RNA 2,500 to 200,000 copies/mL
• Documented exposure to at least two different HAART regimens containing NRTI(s), NNRTI(s), and at least two (2) PIs, excluding low dose Ritonavir, for a minimum of four months each OR documented three class resistance by genotype and/or phenotype AND in the opinion of the investigator, have few if any effective treatment options available.
Four cohorts of 10 subjects each were randomized in a ratio of 4:1 to KP-1461 or placebo, respectively. Successive dose cohorts received 400 mg, 800 mg and 1600 mg of KP-1461 every 12 hours. An additional 10 subjects were enrolled at the 1600-mg dose for a total of 20 subjects at the 1600-mg dose. A fifth cohort of 5 subjects was randomized in a ratio of 4:1 to KP-1461 or placebo, respectively, at a KP-1461 dose of 3200 mg every 12 hours. Data on this cohort is not yet complete.
Subjects were off antiretroviral drugs for at least two weeks prior to the first dose of study drug. Each dose was taken on an empty stomach. Subjects in each cohort were treated for 14 days with an additional 14 days of follow-up. Entry into the next higher dose cohort began when the previous cohort completed enrollment and ≥ 80% of subjects completed dosing plus 1 week of observation. Approval to dose escalate was provided by a Safety Review Committee (SRC). If two or more subjects in a cohort experienced a Grade 3 or 4 toxicity, the SRC reviewed the event to determine whether further dose escalation was safe. If the SRC halted escalation, the previous dose level would be considered the maximum tolerated dose (MTD). Blood was collected for a 12-hour PK profile after the first dose and a 24-hour PK profile after the last dose of KP-1461 (Day 14) and at Days 8, 16, 21 and 28 to determine trough drug concentrations. Routine safety assessments were measured at baseline and on Days 4, 6, 8, 10, 12, 14, 16, 21, and 28. Follow-up safety evaluations occurred at Days 16, 18, 21 and 28. Subjects were able to restart antiretroviral medications after the Day 28 visit. If restarted within 2 weeks of the Day 28 visit, blood was to be collected for
viral load and CD4+ count 84 days afterward, if subject agreed to do so.
This study was conducted in accordance with the clinical research guidelines defined in the U.S. 21 CFR Parts 50, 56, and 312, the principles enunciated in the World Assembly Declaration of Helsinki and its most recent amendments, and the principles defined by the International Conference on Harmonization.
References
Overbaugh J and Bangham CR. Selection Forces and Constraints on Retroviral Sequence Variation. Science 2001; 292(5519):1106-1109.
Eigen M. Error Catastrophe and Antiviral Strategy. Proc Natl Acad Sci USA 2002; 99(21):13374-6.
Anderson JP, Daifuku R, and Loeb LA. Viral Error Catastrophe by Mutagenic Nucleosides. Annu Rev Microbiol 2004; 58:183-205.
Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, and Mullins JI. Lethal Mutagenesis of HIV with Mutagenic Nucleoside Analogs. Proc Natl Acad Sci USA 1999; 96(4):1492-1497.
Harris KS, Brabant W, Styrchak S, Gall A, Daifuku R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antiviral Research 2005;67:1-9.
|
| |
|
|
|
|
|