 |
|
|
| |
CXCR4-using Virus Detected in Patients Receiving Maraviroc in the Phase 3 studies MOTIVATE 1 and 2 Originates from a Pre-existing Minority of CXCR4-using Virus
|
| |
| |
Reported by Jules Levin
XVI International HIV Drug Resistance Workshop. Barbados, 12-16 June 2007
Marilyn Lewis1, Paul Simpson1, Signe Fransen2, Wei Huang2, Jeannette Whitcomb2, Michael Mosley1, David L Robertson3, Roy Mansfield1,
Giuseppe Ciaramella1 and Mike Westby1
1Pfizer Global Research and Development, Kent, UK 2Monogram Biosciences, California, USA 3University of Manchester, Greater Manchester, UK
AUTHOR CONCLUSIONS
--Intensive clonal analyses support the emergence of CXCR4-using viruses as being a consequence of selective suppression of R5 viruses by MVC.
--This is supported by the reversion to R5 in those patients with subsequent off-drug follow-up.
-- A similar fluctuation in ARV- sensitive and resistant virus subpopulations has been seen during and following treatment with enfuvirtide (within 16 weeks)12 and with lamivudine (loss of M184V within 12-36 weeks)13.
ABSTRACT 56
CXCR4-using virus detected in patients receiving maraviroc in the Phase III studies MOTIVATE 1 and 2 originates from a pre-existing minority of CXCR4-using virus
M Lewis1, P Simpson1, S Fransen2, W Huang2, J Whitcomb2, M Mosley1, DL Robertson3, R Mansfield1, G Ciaramella1 and M Westby1
1Pfizer Global Research and Development, Kent, UK
2Monogram Biosciences, California, USA
3University of Manchester, Greater Manchester, UK
INTRODUCTION: Maraviroc in combination with optimized background therapy (OBT) has demonstrated significantly greater virological suppression compared to OBT alone in two double-blind clinical studies of treatment-experienced patients with CCR5-tropic virus. This investigation aimed to understand whether CXCR4-using viruses detected in some patients emerged from a pre-treatment CXCR4-using reservoir, or as a result of mutation from a CCR5-tropic progenitor whilst on treatment ('tropism switch').
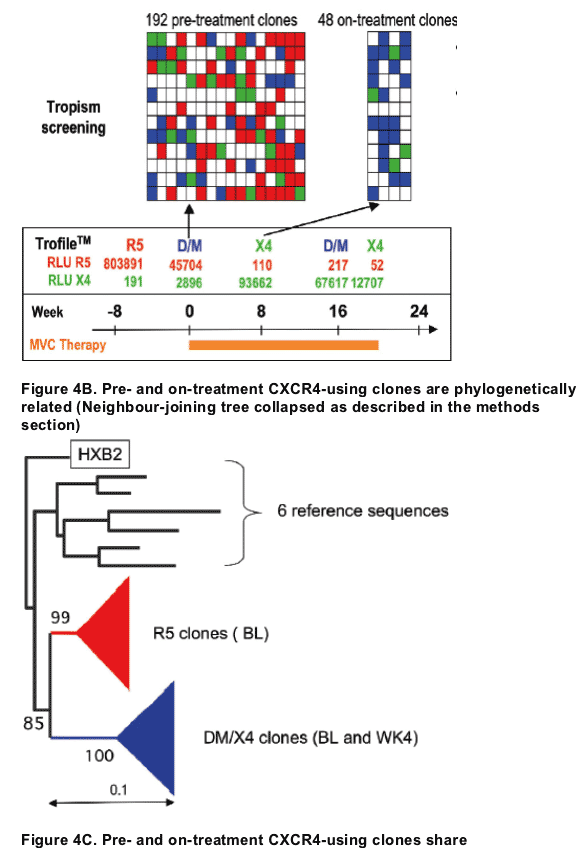
METHODS: Detailed clonal analyses were conducted on samples from 20 patients (16 on maraviroc and 4 on placebo) in whom CXCR4-using virus was detected by the Trofile assay. For each patient, HIV-1 envelope (Env) clones were amplified from plasma: 192 baseline and 48 on-treatment clones were then randomly selected and phenotypically screened for tropism. Env sequences
from these clones were aligned and phylogenetic trees were constructed.
RESULTS: For 14 patients, CXCR4-using env clones identical/similar in sequence to the on-treatment CXCR4-using virus were identified in the baseline sample. The CXCR4-using clones were present at baseline at a low frequency
(1-6%) in 10 of 14 patients, whilst the baseline samples of 4 patients had >10% of CXCR4-using clones; the latter being consistent with the D/M-tropic assignment of their plasma sample. For the remaining six patients, the on-treatment CXCR4-using clones were phylogenetically distinct from baseline and on-treatment CCR5-tropic clones, and contained between seven and 17 amino acid differences in the 35-amino acid V3 loop alone. The origin of on-treatment CXCR4-using virus did not differ between patients receiving maraviroc or placebo, but the clonal screening showed an almost complete loss of CCR5-tropic clones in the on-treatment samples from the patients receiving maraviroc. Of the 20 patients, six continued to respond to therapy at week 24 (4 on maraviroc and 2 on placebo).
CONCLUSIONS: This detailed clonal analysis supports a pre-existing CXCR4-using virus as the most likely origin of on-treatment CXCR4-using virus. No evidence for coreceptor switching was identified whilst on therapy. The apparent loss of CCR5-tropic clones in the on-treatment samples is consistent with their strong in vivo inhibition by maraviroc.
Background & Objectives
Maraviroc (MVC) is a CCR5 antagonist in Phase 2b/3 clinical development for the treatment of HIV-1 infection. MVC in combination with optimized background therapy (OBT) has demonstrated significantly greater virologic suppression compared to OBT alone in two double-blind clinical studies of treatment experienced patients with CCR5-tropic (R5) virus.1,2 Virus populations that exist in HIV-1-infected patients may comprise pure populations of R5 viruses, CXCR4-tropic viruses, dual-tropic viruses, or mixed populations of two or more of the above. MVC is not active against X4 virus, dual-tropic virus or mixed-tropic virus (collectively termed CXCR4-using virus). The effect that the use of a CCR5 antagonist will have on viral tropism in infected individuals remains uncertain.
In pre-clinical3 and clinical4 studies, resistance to MVC has not been associated with a change in co-receptor tropism (except where this also occurred in control virus passaged in parallel without MVC).
Co-receptor switching frequently involves multiple mutations with a fitness cost incurred by transitional intermediates.5
In two (of 64) patients with R5 HIV-1 who received MVC as monotherapy for 10 days, viral load declined during treatment and CXCR4-using virus was detected at Day 11.6
-- Phylogenetic analysis indicated that the CXCR4-using variants probably emerged by outgrowth of a pre-treatment CXCR4-using reservoir, rather than via co-receptor switch.
-- Circulating virus reverted to predominantly R5 following cessation of MVC.
-- This was consistent with a third patient who had D/M virus at Day 1.
Similar findings have been reported from clinical trials of aplaviroc and vicriviroc.7,8
This investigation aimed to understand whether CXCR4-using viruses observed in patients treated with MVC during the MOTIVATE 1 and MOTIVATE 2 clinical trials emerged from a pre-treatment CXCR4-using reservoir, or as a result of mutation from an R5 progenitor ('tropism switch').
Methods
Patients
MOTIVATE 1 and 2 are ongoing, randomized, double-blind Phase 3 trials in antiretroviral (ARV)-experienced patients with only R5 HIV-1 detected at screening (Trofile assay, Monogram Biosciences9). MOTIVATE 1 was conducted in study centres across North America; MOTIVATE 2 was conducted in study centres across Europe, Australia, and North America.
Patients in both trials were randomized to receive placebo or MVC QD or BID (1:2:2). All three treatment groups also received an optimized background therapy (OBT) of 3-6 ARVs (pharmacokinetic boosting doses of retrovir not counted as an ARV).
Patients receiving a protease inhibitor (except tipranavir) and/or delavirdine in their OBT received 150 mg dose of MVC; all other patients received 300 mg dose of MVC.
The duration of both trials is 48 weeks with a planned interim analysis after 24 weeks.
The origins of the patients included in this investigation of tropism changes are detailed in Figure 1.
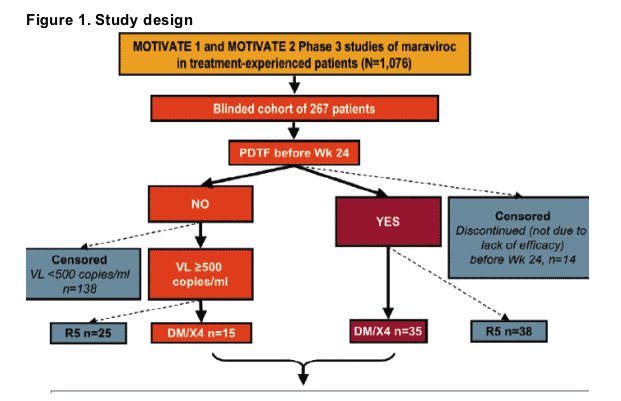
Twenty patients with D/M virus were selected for the tropism sub-study, including:
-- 16 on MVC and 4 on placebo
-- early failures (n=9) who reached protocol-defined treatment failure (PDTF) by Week 8
-- late failures (n=5) who reached a PDTF after Week 8
-- patients (n=6) who did not reach a PDTF by Week 24
-- patients whose virus was classified as R5* at screening but DM/X4* at Day 1 (pre-dose, n=5)
-- patients in whom virus was classified as DM/X4* only after receiving blinded study drug (n=15).
*Tropism as assigned by the Trofile assay, Monogram Biosciences9.
2 patients on treatment samples were N/R
PDTF = Protocol defined treatment failure
Phenotypic and genotypic tropism assignmentL
The Trofileassay9 was used to assign viral tropism during the clinical trials and to confirm the tropism of clones.
Note that Trofile results are reported as R5, X4 or dual/mixed (D/M). Thus dual-tropic clones are assigned as 'D/M'.
For comparison, tropism was also assigned using the 11/2510 rule and the position-specific scoring matrix (PSSM)11.
Screening for minority CXCR4-using variants
The number of functional envelope clones to screen in order to detect with 99% probability, a minority of CXCR4-using variants (at 5%) in a pre-treatment sample was calculated as: log(1-P)/log(1-p), where P = probability of detecting CXCR4-using Env clones and p = proportion of CXCR4-using clones in the total population(Table 1)

Phylogenetic analysis
To compare pre- and on-treatment viruses, V3 alignments and phylogenetic trees were constructed using a modification of previously described methods.3 The following techniques were used:
Alignment of sequences (approx 290 nucleotides long including the V3 loop) with ClustalX using seven reference sequences, HXB2 to root the trees and 6 sequences from 3 other patients to confirm that all viruses in a single patient were related to one another.
Construction of neighbour-joining trees in PHYLIP, bootstrap values were calculated using 100 repititions and shown on the branches
-- neighbour-joining trees edited using the phylogeny programme in MEGA (http://www.megasoftware.net/), collapsing the large branches and annotating accordingly. The size of the triangle is proportional to the number of branches collapsed. Scale bar shown to represent nucleotide substitutions per site.
Construction of maximum parsimony trees in PHYLIP (version 3.65, http://evolution.genetics.washington.edu/phylip.html).
Construction of maximum likelihood trees using PAUP (Sinauer Associated Inc. Sunderland, MA).
RESULTS
Example of a patient where on-treatment CXCR4-using virus shared a common ancestor with pre-treatment clones (Patient T6, virus classified as R5 at baseline) (Figure 2)
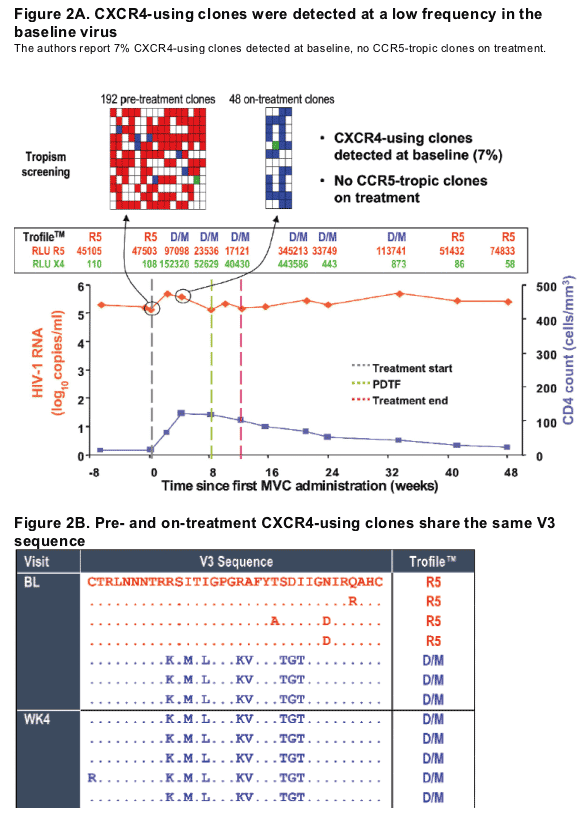
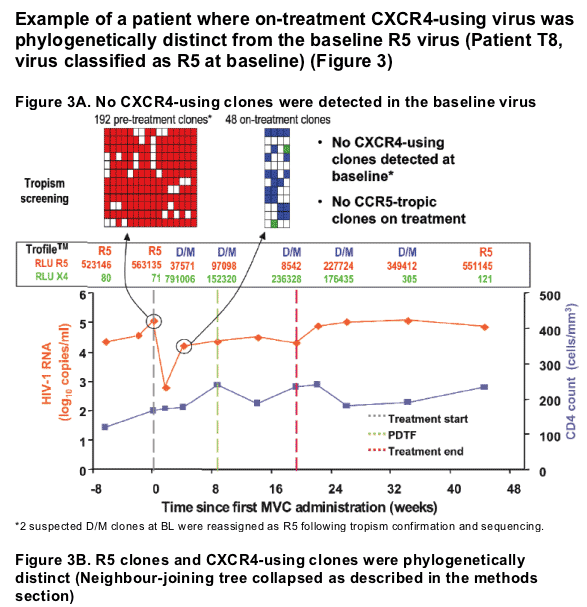
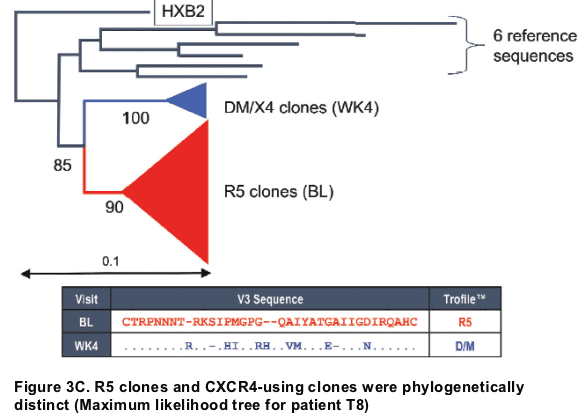
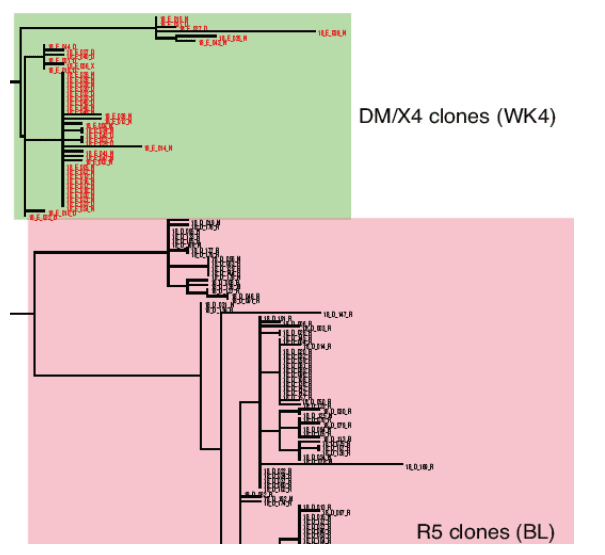
Change in viral tropism on treatment is due to selection of pre-existing CXCR4-using minor variants. (Table 2).
Either
-- CXCR4-using env clones were detected at baseline
or
-- On-treatment CXCR4-using clones were genetically distinct from R5 clones (7-17aa in V3 loop alone).
There were no differences in origin of CXCR4-using virus seen between MVC and placebo patients.
Changes in tropism were seen in the absence of treatment failure.

Overall there was a 90% concordance between tropism assignment of Trofile and PSSM algorithm for individual clones (Table 3). However, the sensitivity of the PSSM or 11/25 algorithms to detect CXCR4-usage was lower than for detection of CCR5-tropism.

REFERENCES
1. Lalezari J, et al. 14th CROI 2007. Presentation 104bLB.
2. Nelson M, et al. 14th CROI 2007. Presentation 104aLB.
3. Westby M, et al. J Virol 2007; 81:2359-2371.
4. Mori J, et al. XVI International HIV Drug Resistance Workshop. Barbados, June 12 16 2007. Abstract S12.
5. Pastore C, et al. J Virol 2004; 78:7565-7574.
6. Westby M, et al. J Virol 2006; 80:4909-4920.
7. Kitrinos K, et al. Antivir Ther 2005; 10:S68.
8. Schurmann D, et al. CROI 2004; Abstract 140LB.
9. Whitcomb JM, et al. Antimicrob Agents Chemother 2007; 51:566-575.
10. DeJong JJ et al. J Virol. 66:6777-6780.
11. Jensen MA et al. J Virol. 77:13376-13388.
12. Deeks S, et al. J Inf Dis 2007; 195: 387-391.
13. Deeks S, et al. J Inf Dis 2005; 192:1537-1544.
|
| |
|
|
|
|
|