 |
|
|
| |
Antiviral Activity of the HCV Polymerase Inhibitor PF-00868554 Administered as Monotherapy
in HCV Genotype 1 Infected Subjects
|
| |
| |
Reported by Jules Levin
AASLD Nov 3 2008 San Francisco, CA
Jennifer L. Hammond1, Maria C. Rosario1, Frank Wagner2, Dago Mazur3, Constantino Kantaridis4, Vivek S. Purohit1, L. Kathryn Durham1, Shyla Jagannatha1, Michael F. DeBruin1
1Infectious Diseases, Pfizer Global Research and Development, New London, CT, USA; 2Charite Research Organisation, Berlin, Germany; 3Clinical Pharmacology, Parexel International, Berlin, Germany;
4Pfizer Clinical Research Unit, Pfizer Global Research and Development, Brussels, Belgium
Author Conclusions
PF-00868554 potently inhibited viral replication in a dose-dependent manner in treatment-naïve subjects infected with HCV genotype 1. Mean maximum reductions in plasma HCV RNA levels ranged from -0.97 to -2.13.
The PK of PF-00868554 in treatment-naïve HCV-infected subjects was similar to that of healthy volunteers.1
PF-00868554 has a good safety profile and is well tolerated at 100-450 mg BID and 300 mg TID in HCV genotype 1 infected subjects.
Results from the present study support the further evaluation of PF-00868554 - a study investigating PF-00868554 in combination with pegylated interferon alpha-2a and ribavirin in treatment-naïve subjects is currently underway.
Abstract
BACKGROUND: PF-00868554 is a novel, potent, non-nucleoside inhibitor of the HCV polymerase. PF-00868554 inhibits genotype 1a and 1b replicons in vitro, with an overall mean EC50 of 0.059 _M. The safety and tolerability of PF 00868554 has been demonstrated in healthy volunteers administered PF-00868554 up to 300 mg TID for 14 days. The objectives of this study were to evaluate the safety, tolerability, pharmacokinetics (PK) and antiviral activity of PF-00868554 in HCV-infected subjects.
METHODS: This was a randomized, double-blind, placebo-controlled, sequential group study. Subjects eligible for participation were those naïve to previous interferon-based HCV therapy and infected with a genotype 1 strain of the virus. Four cohorts of eight subjects were randomized (6:2) to receive oral doses of PF-00868554 (100, 300 and 450 mg BID and 300 mg TID) or placebo, for 8 days.
RESULTS: Of the 32 subjects randomized, the majority were male (84%) and Caucasian (97%). Mean baseline HCV RNA levels ranged from 5.8 to 6.1 log10 IU/mL. All doses of PF-00868554 were well tolerated. The most frequently reported all-causality adverse events (AEs ) were headache, flatulence, and fatigue. All AEs were mild or moderate in severity. There were no dose-limiting AEs, Grade 3 or 4 laboratory abnormalities, withdrawals due to AEs, serious AEs, or deaths. The half life of PF-00868554 in this study ranged from 8 to 12 hours. All doses achieved plasma concentrations that exceeded the median protein binding adjusted in vitro EC50 value for genotype 1 HCV throughout the duration of the dosing interval.
The mean maximum reduction (log10) in HCV RNA achieved with PF-00868554 100, 300 or 450 mg BID or 300 mg TID was -0.97, -1.84, -1.74, and -2.13, respectively. The mean reduction in HCV RNA at the end of PF-00868554 treatment on Day 8 was -0.68, -1.26, -1.21, and -1.95, respectively. HCV RNA decreased rapidly during the first 48 hours of PF-00868554 treatment. Following this first phase of viral suppression, most subjects experienced a plateau or rebound in HCV RNA; however, some subjects' HCV RNA continued to decline through the completion of dosing on Day 8.
CONCLUSIONS: Results from this study indicate that PF-00868554 was well tolerated at all dose levels. PF-00868554 potently inhibited viral replication in HCV-infected, treatment-naïve subjects, with mean maximum reductions in HCV RNA ranging from -0.97 to -2.13. Results from the present study support the further evaluation of PF-00868554, and a study investigating PF-00868554 in combination with pegylated interferon alpha-2a and ribavirin in treatment-naïve subjects is currently underway.
Background
PF-00868554 is a potent and selective non-nucleoside inhibitor of the HCV polymerase enzyme with a mean IC50 of 0.015 _M against genotype 1 polymerase.
PF-00868554 binds at the "Thumb 2" site of the HCV polymerase enzyme (Figure 1).
PF-00868554 has potent in vitro antiviral activity with an overall mean EC50
against genotype 1 replicons of 0.059 _M. It appears to be equipotent against
both genotypes 1a and 1b.
In a study of healthy non-HCV infected volunteers, multiple oral doses of PF-00868554 up to 300 mg TID demonstrated a good safety profile and were well tolerated over 14 days.1 In addition, doses of PF-00868554 ≥100 mg BID achieved plasma concentrations expected to inhibit viral replication in vivo.
In this dose-escalating study we report the antiviral activity, PK, safety and
tolerability profile of PF-00868554 in treatment-naïve HCV-infected subjects.
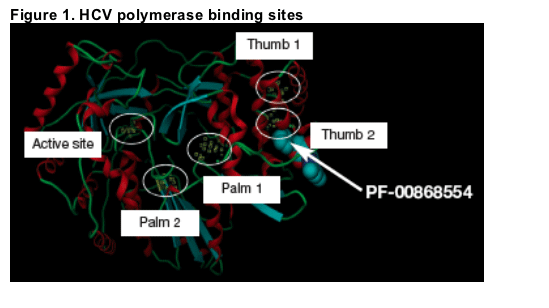
Objectives
Primary
To assess the safety and tolerability of multiple oral doses of PF-00868554 in HCV genotype 1 infected subjects.
To assess the PK of multiple oral doses of PF-00868554 in HCV genotype 1
infected subjects.
Secondary
To evaluate the effect of multiple oral doses of PF-00868554 on plasma HCV RNA levels in subjects chronically infected with HCV genotype 1.
Methods
Study design
Double-blind, randomized, placebo-controlled, sequential dose escalation study.
The protocol was reviewed and approved by an independent ethics committee
and all subjects gave written informed consent.
A total of 32 HCV-infected subjects were assigned to 1 of 4 treatment cohorts
(8 subjects per cohort: 6 active and 2 placebo).
Subjects received PF-00868554 or placebo, administered as an oral solution
under fasted conditions for 8 days as follows:
- Group 1: 100 mg PF-00868554/placebo BID on Days 1-7 and a single AM dose on Day 8
- Group 2: 300 mg PF-00868554/placebo BID on Days 1-7 and a single AM dose on Day 8
- Group 3: 450 mg PF-00868554/placebo BID on Days 1-7 and a single AM dose on Day 8
- Group 4: 300 mg PF-00868554/placebo TID on Days 1-7 and
a single AM dose on Day 8. Key inclusion criteria
Male and female (of non-childbearing potential) subjects between the ages of 18 and 65 years.
A diagnosis of chronic HCV genotype 1 infection (HCV RNA detectable in serum) for at least 6 months.
HCV RNA ≥100,000 IU/mL at screening.
Key exclusion criteria
_ HBV or HIV coinfection.
_ Current or prior treatment with interferon and/or ribavirin.
_ Prior treatment with an investigational anti-HCV agent exceeding 2 weeks' duration.
_ Evidence of severe or decompensated liver disease.
_ Liver disease unrelated to HCV-infection.
Antiviral activity assessments
_ The antiviral activity of PF-00868554 was assessed by measuring HCV RNA levels over time. Plasma HCV RNA levels were quantified using the Abbott RealTime HCV assay (LLOQ = 12 IU/mL) at the following time points:
- Screening
- Day 0
- Days 1, 2 and 3: samples were taken at 0 hours (pre-dose) and every 6 hours up until 48 hours post-dose
- Days 4, 5, 6, 7 and 8: samples were taken pre-dose every 24 hours
- Days 10, 12, 14, 21 and 28 (Follow up visit): samples were taken in the morning.
Pharmacokinetic assessments
Full PK profiles for PF-00868554 were obtained from samples obtained on Days 1 and 8. Pre-dose samples were collected on Days 2 through 7. Additionally, on Day 8, samples were collected up to 48 hours following the last dose.
Plasma concentrations of PF-00868554 were determined by a validated high performance liquid chromatography tandem mass spectrometry method (HPLC-MS/MS) with a lower limit of quantification of 2 ng/mL.
Safety and tolerability assessments
Physical examinations, electrocardiograms (ECGs), vital signs measurements and clinical laboratory tests were performed and adverse events (AEs) were monitored throughout the study to assess the safety and tolerability of PF-00868554.
Statistical analysis
PK and statistical analyses were conducted using WinNonlin Professional
version 5.2 (Pharsight Corporation, CA, USA).
Results
Subject demographics and baseline characteristics
All 32 subjects who were randomized completed the study.
There were no notable differences in demographic parameters and baseline HCV RNA levels between subjects who received active treatment and those who received placebo (Table 1).
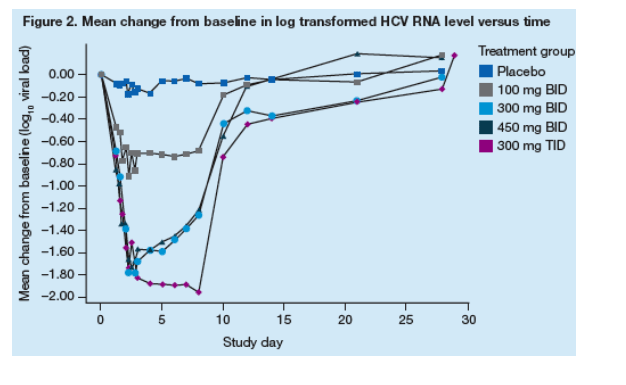
Viral inhibition
HCV RNA levels decreased rapidly during the first 48 hours of PF-00868554 administration (Figure 2).
Following this first phase of viral suppression, most subjects experienced a plateau or rebound in HCV RNA; however, some subjects' HCV RNA continued to decline through the completion of dosing on Day 8 (Figure 2).
One subject in the 300 mg BID group and 3 subjects in the 300 mg TID group exhibited sustained suppression of HCV viremia through completion of dosing on Day 8.
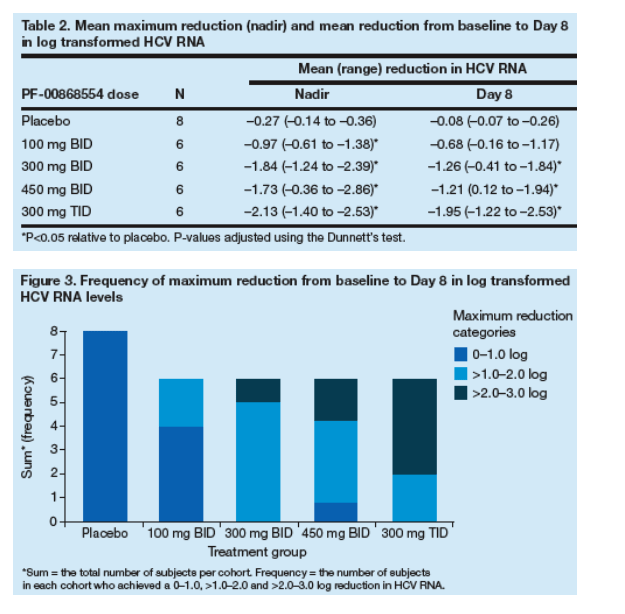
Mean maximum reductions in HCV RNA levels ranged from -0.97 (100 mg BID) to -2.13 (300 mg TID). On Day 8, the mean reduction in HCV RNA from baseline ranged from -0.68 (100 mg BID) to -1.95 (300 mg TID) (Table 2).
One subject in the 450 mg BID cohort experienced a <0.5 log reduction in HCV RNA concentrations relative to baseline. Sequence analysis of the NS5B gene of the baseline virus from this subject identified an R422K mutation. As residue 422 is located adjacent to the PF-00868554 binding pocket, the R to K substitution could potentially reduce susceptibility to PF-00868554 (to be confirmed in future studies). If results from this subject were excluded from the analysis, the mean reduction in HCV RNA for the 450 mg BID cohort at nadir and Day 8 were -2.01 and -1.48, respectively.
A total of 0/6, 1/6, 2/6 and 4/6 subjects treated with PF-00868554 in the 100 mg BID, 300 mg BID, 450 mg BID and 300 mg TID groups experienced a >2.0 log maximum reduction in HCV RNA concentrations from baseline to Day 8 (Figure 3).
Pharmacokinetics
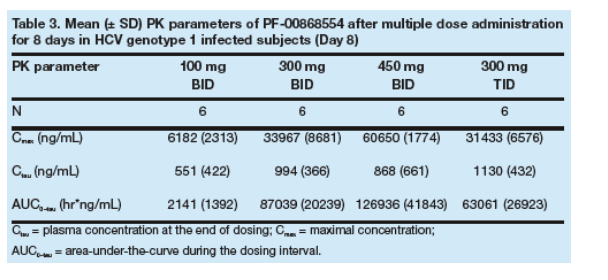
Mean Cmax and AUC for PF-00868554 increased with increasing dose (Table 3).
PF-00868554 exhibited non-linear PK across all doses studied. Increases in both mean AUC and Cmax were greater than the proportional increases in PF-00868554 dose (Table 3).
Tmax was 0.5 to 1.5 hours post-dose.
The half-life of PF-00868554 ranged from 10-12 hours for all dosing groups and therefore supported BID dosing (Table 3).
Safety and tolerability
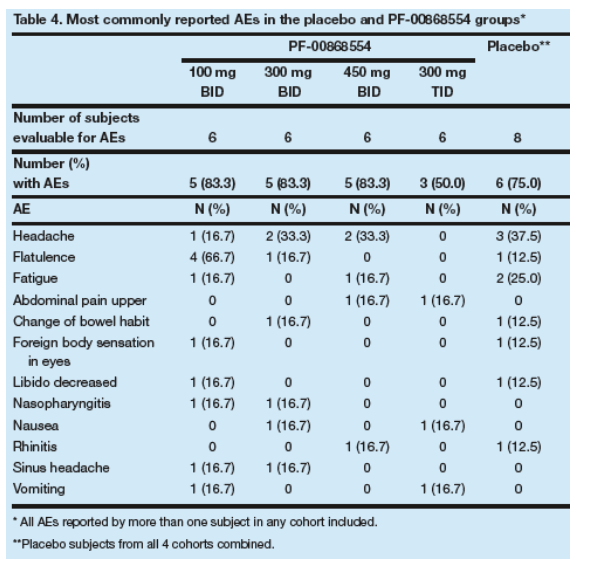
All AEs were mild or moderate in intensity.
The most frequently reported AEs were headache, flatulence and fatigue (Table 4).
There were no deaths, serious AEs, dose reductions, temporary discontinuations or withdrawals due to AEs reported in this study.
There were no trends in laboratory abnormalities and none of the laboratory abnormalities observed during the study exceeded Grade 2.
There were no clinically significant changes in vital signs or ECG parameters.
References
1. Hammond JL, et al. Safety, tolerability and pharmacokinetics of the HCV polymerase inhibitor PF-00868554 following multiple dose administration in healthy volunteers. Poster 1898 presented at AASLD 2008.
|
| |
|
|
|
|
|