 |
|
|
| |
Long-Term Safety of Vicriviroc: out to 4 yrs
|
| |
| |
Reported by Jules Levin
ICAAC/IDSA Oct 28 208 Wash DC
Dunkle, WL, Greaves, MC, MC McCarthy, E Lee, N Case, J Shen, and C Mak, Schering-Plough Research Institute, Kenilworth, NJ, USA
AUTHOR CONCLUSIONS
These data represent the longest treatment duration and clinical experience reported to date for a CCR5 antagonist.
This analysis of 205 subjects demonstrates that long-term success with vicriviroc (VCV) treatment results from sustained virologic suppression and elevated CD4 counts with no clearly associated adverse events.
In this study VCV as part of an antiretroviral regimen containing a PI/r, provides durable suppression of HIV and elevation of CD4 cells.
In this study VCV therapy had excellent tolerability and is not associated with hepatotoxicity, seizures, or ischemic cardiovascular events.
VCV (30 mg QD), as part of an antiretroviral regimen containing a PI/r, is now being studied in fuly-enrolled Phase III clinical trials of HIV-monoinfected and HCV/HIV-coinfected or HBV/HIV-coinfected treatment-experienced patients.
Abstract Results
Background: Vicriviroc (VCV) is a next-generation CCR5 antagonist with potent in vitro anti-HIV activity and sustained antiviral efficacy in treatment-experienced subjects receiving a PI/r-containing ART regimen. Over 200 subjects received VCV in Phase II. The long-term safety of VCV was evaluated.
Methods: Open-label VCV was provided to subjects at the completion of 48 weeks in studies ACTG5211 and VICTOR-E1. VCV was initiated at doses ranging from 5-30 mg, but over time all subjects escalated to 30 mg QD, as safety and efficacy supported that dose. Data were pooled for all subjects, regardless of initial dose, as no safety differences were seen across the doses. AIDS complications, malignancies, infections and liver-related abnormalities were evaluated specifically. Changes in HIV RNA and CD4 counts were based on available data at defined intervals.
Results:
205 subjects with advanced HIV infection were enrolled in the 2 trials; 196 received >12 weeks VCV treatment.
Mean duration of Tx for all 205 was 96 weeks (range 1-216 weeks).
AIDS complications included 9 OIs, 2 KS, 2 wasting syndrome and 1 PML. 13 malignancies were reported: 7 skin (including the 2 KS), 5 lymphomas,
1 gastric carcinoma. Liver abnormalities included: 1 cirrhosis; 1 hepatosplenomegaly; 14, 17 and 25 elevated ALT, AST and bilirubin, respectively. AST/ALT elevations were generally mild and deemed not drug-related. All hyperbilirubinemia was associated with atazanavir (without AST/ALT elevation). Infections reported in 35% of subjects included sinusitis (11%), bronchitis (9%), URI (9%), herpes simplex (6%), influenza (5%), and pneumonia (5%).
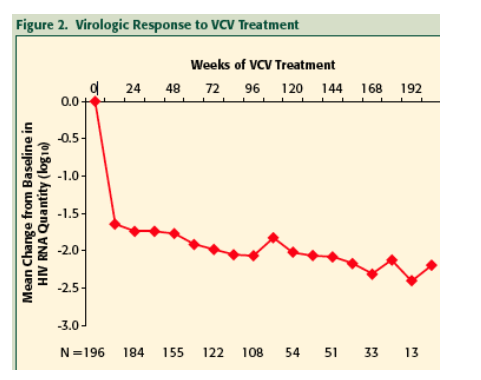
Median change from baseline HIV RNA was -2.1, -2.2, -2.3, -2.3 (log10), and mean change from baseline CD4 was +121, +144, +158 and +143 (cells/mL) at 48, 96, 144 and 168 weeks, respectively.
Conclusions: These subjects represent the longest clinical experience and treatment duration with a CCR5 antagonist reported to date. VCV demonstrated excellent tolerability, few HIV complications and no clearly VCV-related toxicity. Sustained antiviral effect with prolonged maintenance of improved CD4 counts was observed.
Introduction
VCV is a next-generation extracellular inhibitor of HIV infection designed to block entry of infectious virions into uninfected CD4+ cells via antagonism of the CCR5 coreceptor (CCR5 antagonist).1
VCV is highly orally bioavailable and has a long plasma half-life, allowing for once daily dosing with or without food.2,3
VCV plasma concentrations are increased by co-administration of inhibitors of
CYP3A4 (e.g. ritonavir)4, and higher plasma concentrations (as measured by Cmin) correlate with greater virologic suppression.5
VCV does not require dose adjustment when combined with other antiretrovirals as part of a PI/r-containing regimen.
VCV has strong potential as a new treatment option for HIV-infected patients and is now in Phase III clinical trials in treatment-experienced subjects (30 mg QD as part of a PI/r-containing regimen).
Background
Efficacy: Previously presented clinical trial data have demonstrated a sustained and durable antiretroviral effect in treatment-experienced HIV-infected subjects taking VCV in combination with a PI/r-containing OBT.
Phase II Trial ACTG52116
--VCV-treated subjects experienced greater declines in HIV RNA levels, and a greater proportion achieved virologic suppression at 24 and 48 weeks, as compared with OBT alone.
--VCV-treated subjects experienced fewer virologic failures as compared with
OBT alone.
--VCV-treated subjects experienced increased CD4 cell counts at 24 and 48 weeks as compared with OBT alone.
--A dose-response was evident: the 10 mg and 15 mg QD doses of VCV were
more effective than the 5 mg QD dose, but no apparent difference between
10 and 15 mg.
Phase II Trial P03672 (VICTOR-E1)7
--VCV-treated subjects experienced greater declines in HIV RNA levels, and a
greater proportion achieved virologic suppression at 48 weeks, as compared
with OBT alone.
--VCV-treated subjects experienced increased CD4 cell counts at 48 weeks as
compared with OBT alone.
--VCV plasma concentrations correlated with virologic suppression: 58% vs. 27% of subjects achieved <50 copies/mL when Cmin was >100 ng/mL vs.
--Trends toward superiority of the 30 mg vs. 20 mg QD dose were observed in
subjects with >100,000 copies/mL HIV-1 RNA and in subjects with
--Few VCV-treated subjects experienced virologic failure, which was not
necessarily associated with the detection of DM/X4 variants.
Rollover Trial P04100 (open-label continuation of subjects in ACTG5211)8
--Declines in HIV RNA levels and full virologic suppression achieved in ACTG5211 were sustained for 18 and up to 24 months.
--Increased CD4 cell counts achieved in ACTG5211 were sustained for 18 and
up to 24 months.
--AIDS-related opportunistic infections not observed.
--Few VCV-treated subjects experienced virologic failure, which was not necessarily associated with the detection of DM/X4 variants or VCV resistance.
Safety: Previously presented data from these same three trials6-8 have observed no signature toxicities associated with VCV treatment.
No attributable hepatotoxicity:
-- No elevated liver enzymes.
-- No elevated bilirubin.
No attributable cardiovascular toxicity:
-- No ischemic events.
No attributable CNS toxicity:
-- No seizures.
Malignancies similar to those reported in ACTG5211 were not observed in either P04100 or P03672 (VICTOR-E1).
Study Objectives
To evaluate the efficacy and safety of long-term VCV-containing HAART in subjects infected with HIV.
This report presents pooled long-term efficacy and safety data from the continuation of P04100 and the rollover of subjects from P03672 (VICTOR-E1).
Study Subjects and Methods
Subjects enrolled in Phase II trial ACTG5211 were continued on open-label VCV with OBT in rollover trial P04100. Subjects enrolled in Phase II trial P03672 (VICTOR-E1) were also given the opportunity to continue open-label VCV with OBT after 48 weeks of blinded therapy. Data from these two continuing cohorts were combined for long-term follow-up and analysis.
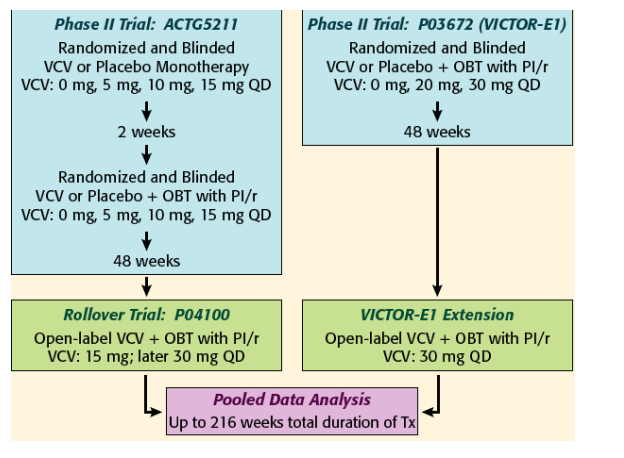
Similarities between the ACTG5211/P04100 and P03672 (VICTOR-E1) subject populations.6-8
-- HIV-infected adults with a history of failing standard 3-drug HAART.
-- R5-tropic HIV only, as detected by first-generation Trofile assay.
-No active AIDS-defining illnesses and generally good health.
-- Randomization stratified by use of enfuvirtide in OBT.
Differences between the ACTG5211/P04100 and P03672 (VICTOR-E1) subject
populations.6-8
-- ACTG5211: Randomization stratified by CD4 counts above or below 50 cells/mm3.
-- P3672 (VICTOR-E1): Randomization stratified by HIV RNA quantities above or
below 100,000 copies/mL.
-- HBV or HCV coinfection with stable liver disease and liver enzymes
All VCV doses escalated to 30 mg QD during open-label VCV treatment, as supported by efficacy and safety analyses of the Phase II trial data.6-8
The composition of the OBT was selected by each subject's provider based upon available genotypic and phenotypic drug resistance test results and was required to contain a PI/r.
Monitoring was done on Days 1, 14, 28, and Month 2, and then every 2-3
months. Parameters monitored for this study included:
-- HIV viral load.
-- CD4 count.
-- AIDS-associated opportunistic infections and conditions.
-- Other infections.
-- Liver-related abnormalities.
-- Malignancies.
-- Viral tropism (data not presented).
RESULTS
Characterization of the combined long-term VCV treatment subject population
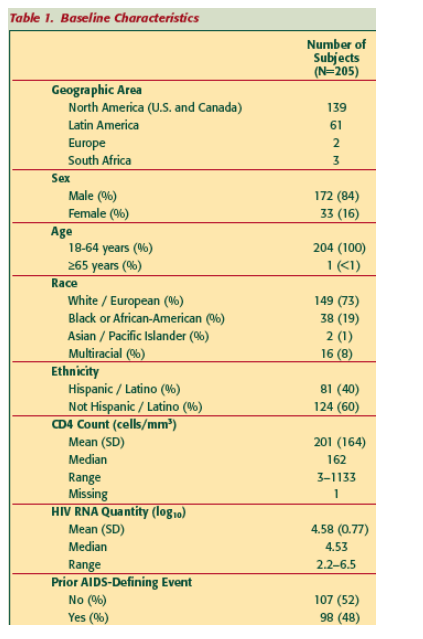
Distribution of VCV treatment duration: VCV-treated subjects (N=205) include those who initiated VCV treatment during a Phase II trial (ACTG5211 and P03672/VICTOR-E1) and those who initiated VCV treatment during rollover trial P04100 or the VICTOR-E1 Extension after having previously received either placebo or no study treatment.
Efficacy analyses were performed on the data from all subjects who completed >/=12 weeks of VCV treatment (N=196), including those who received VCV in one of the placebo-controlled Phase II trials and then continued with open-label treatment, and those who initiated VCV open-label treatment at the completion of each Phase II trial. The data for all subjects receiving VCV were pooled for analysis regardless of their original starting dose of VCV. The number of subjects initially receiving the suboptimal dose of 5 mg QD before escalating to 15 mg QD was too small to impact the overall results.
The virologic response to VCV treatment was evidenced within 12 weeks by a significant decrease in the quantity of HIV RNA detectable in the blood, and this response was sustained for up to nearly two years with continued VCV treatment.
The immunologic response to VCV treatment was evident within 12 weeks by a significant increase in the number of CD4+ cells, and this positive response was sustained for up to nearly two years with continued VCV treatment.
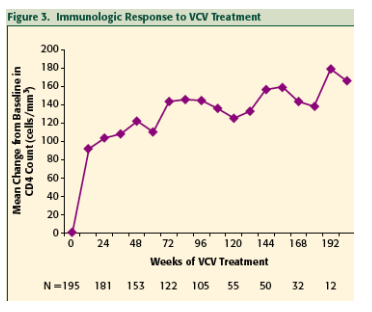
Safety analyses were performed on the data from all subjects who received any VCV treatment (N=205).
Treatment Emergent Adverse Events: AIDS-associated opportunistic infections and conditions were observed infrequently and sporadically. Infections involving the upper and lower respiratory tract were the only other infections or adverse events that occurred in 35% of subjects. Elevations of liver enzymes and bilirubin were noted but judged to be not related to VCV. These infections and other adverse events observed in these study subjects are consistent with expectations for the HIV/AIDS subject population.
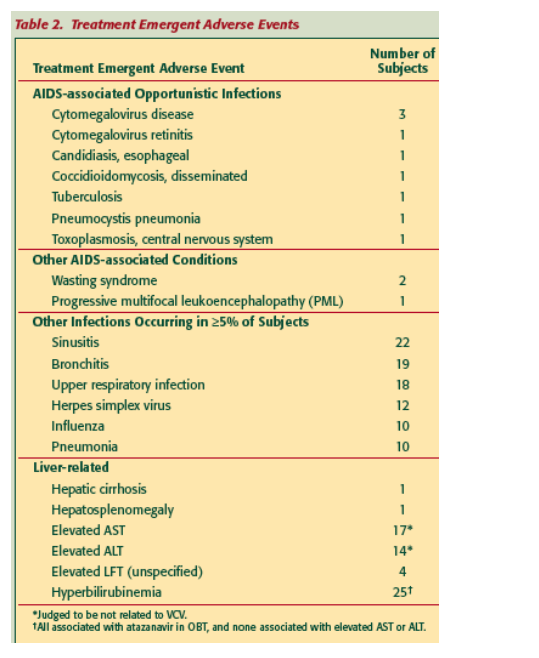
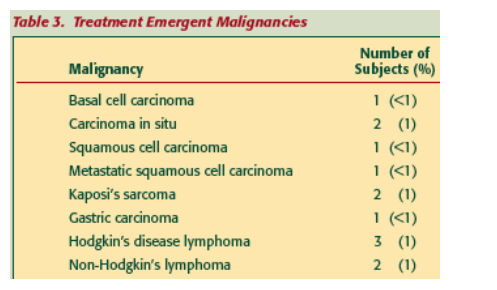
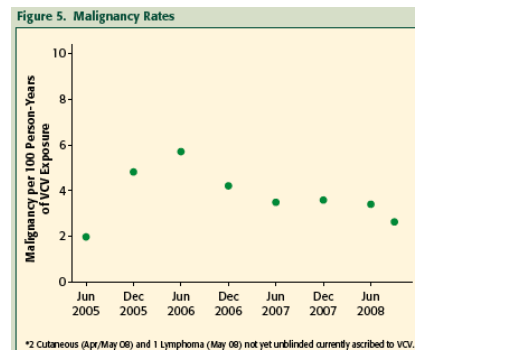
Treatment Emergent Malignancies: The malignancies observed were largely the oncogenic-virus-associated types known to occur with increased frequency in patients with HIV/AIDS and include those initially reported from ACTG5211.
The incidence of malignancy in all VCV study subjects including the Phase 3 studies to date has not increased over time even as the cumulative exposure to VCV by study subjects has increased exponentially.
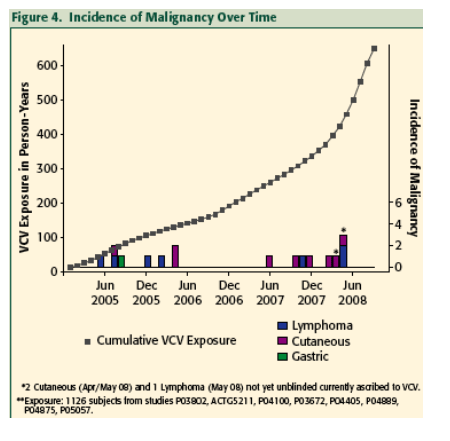
The incidence rates of all malignancies, adjusted for VCV exposure in all study subjects to date, has not increased over time. This suggests that VCV treatment likely does not increase the already elevated risk of developing malignancy in this subject population with HIV/AIDS.
References
1. Strizki JM, Tremblay C, Xu S, et al. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human
immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:
4911-4919
2. Seiberling M, Kraan M, Keung A, et al. Similar increase in SCH 417690 plasma exposure with coadministration of varying doses of ritonavir in healthy
volunteers. In: Program and abstracts of the 6th International Workshop on Clinical Pharmacology of HIV Therapy; April 28-30, 2005; Quebec City, Quebec,
Canada; Abstract 6.4.
3. Sansone A, Keung A, Caceres M, et al. Effect of food on bioavailability of
SCH 417690 in healthy volunteers. In: Program and abstracts of the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy; December 16-19, 2005; Washington, DC, USA; Abstract A-1200.
4. Sansone A, Keung A, Tetteh E, et al. Pharmacokinetics of vicriviroc are not affected in combination with five different protease inhibitors boosted by ritonavir. In: Program and abstracts of the 13th Conference on Retroviruses and Opportunistic Infections; February 5-8, 2006; Denver, CO, USA; Abstract 582.
5. Keung A, Li C, Fellows K, et al. The pharmacodynamic correlation between vicriviroc plasma levels and virologic suppression. In: Program and abstracts of
the 9th International Workshop on Clinical Pharmacology of HIV Therapy; April 7-9, 2008; New Orleans, LA, USA; Abstract O20.
6. Gulick RM, Su Z, Flexner C, et al. Phase 2 Study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS Clinical Trials Group 5211. J Infect Dis. 2007;196:304-312.
7. Zingman B, Suleiman J, DeJesus E, et al. Vicriviroc, a next generation CCR5 antagonist, exhibits potent, sustained suppression of viral replication in treatment-experienced adults: VICTOR-E1 48-week results. In: Program and abstracts of the 15th Conference on Retroviruses and Opportunistic Infections;
February 3-6, 2008; Boston, MA, USA; Abstract 39LB.
8. Gulick R, Haas D, Collier AC, et al. Two-Year Follow-up of Treatment-Experienced Patients on Vicriviroc (VCV). In: Program and abstracts of the 47th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); September 17-20, 2007; Chicago, IL, USA; Abstract 2060.
|
| |
|
|
|
|
|