 |
|
|
| |
TELAPREVIR IN HEPATITIS C GENOTYPE-1-INFECTED PATIENTS WITH PRIOR NON-RESPONSE, VIRAL BREAKTHROUGH OR RELAPSE TO PEGINTERFERON-ALFA-2A/B AND RIBAVIRIN THERAPY: SVR RESULTS OF THE PROVE3 STUDY
|
| |
| |

EASL April 23-26 2009, Copenhagen
Reported by Jules Levin
M. Manns1, A. Muir2, N. Adda3, I. Jacobson4, N. Afdhal5, J. Heathcote6, S. Zeuzem7, H. Reesink8, N. Terrault9, M. Bsharat3, S. George3, J. McHutchison2, A. Di Bisceglie10
1Medical School Hannover, Hannover, Germany, 2Duke University Medical Center, Durham, NC, 3Vertex Pharmaceuticals, Cambridge, MA, 4Weill Cornell Medical College, New York, NY, 5Beth Israel Deaconess Medical Center/Harvard Medical School, Boston, MA, USA, 6University of Toronto, Toronto, ON, Canada, 7Johann Wolfgang Goethe University Hospital, Frankfurt/Main, Germany, 8Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands, 9University of California San Francisco, San Francisco, CA, 10Saint Louis University School of Medicine, Saint Louis, MO, USA
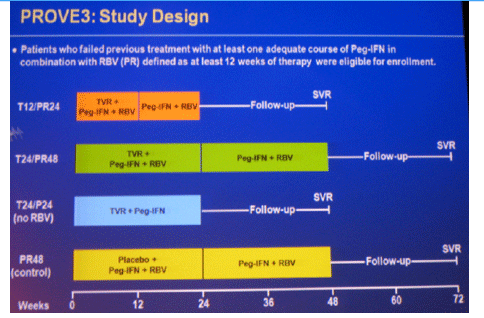
Background: PROVE3 is a randomized, placebo-controlled Phase 2 study assessing safety and efficacy of telaprevir (T) plus Peginterferon-alfa-2a (P) ± Ribavirin (R) in HCV genotype 1 patients who previously failed PR treatment.
Methods: Randomization was 1:1:1:1 to: T/PR for 12-wks, then PR for 12-wks (T12/PR24); T/PR for 24-wks, then PR for 24-wks (T24/PR48); T/P for 24-wks (T24/P24); or placebo/PR (P 180µg/wk, R 1000-1200mg/day) for 24-wks, then PR for 24-wks (PR48).
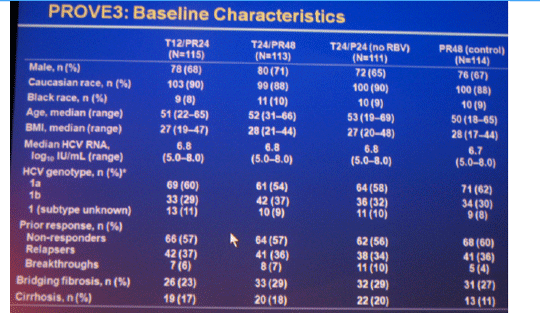
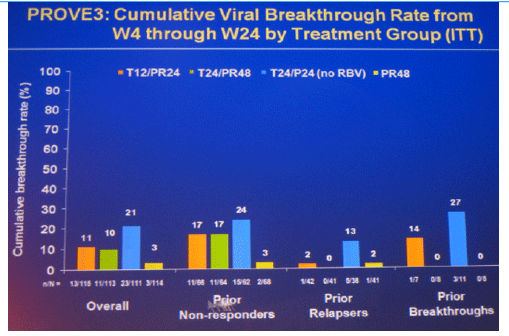
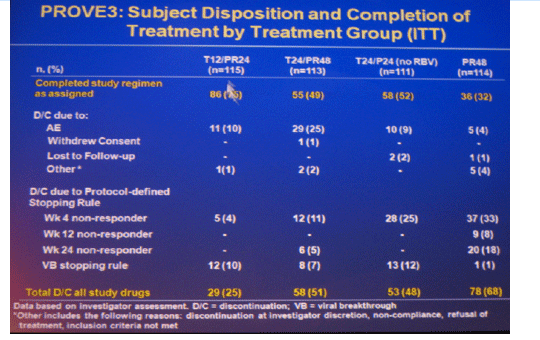
Results: Of 453 patients included in ITT analysis, 418 (92%) had baseline HCV RNA ≥800,000 IU/mL, 196 (43%) had cirrhosis or bridging fibrosis and 40 (9%) were black; 235 (52%) patients completed assigned treatment.
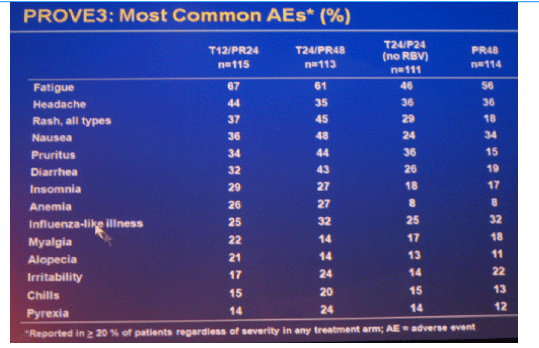
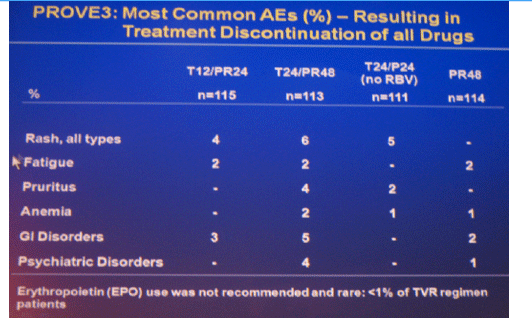
The most frequent adverse events that occurred with a greater incidence in T12/PR24 or T24/PR48 than PR48 were fatigue, nausea, headache, rash, pruritus, diarrhea, anemia, insomnia, pyrexia, alopecia, and chills. Grade 3 rash was observed in 7 (6%), 5 (4%), 6 (5%) and 1 (1%) patients in T12/PR24, T24/PR48, T24/P24, and PR48, respectively. Grade 3 anemia was observed in 0 (0%), 7 (6%), 1 (1%) and 1 (1%) patients in T12/PR24, T24/PR48, T24/P24 and PR48, respectively. Eleven (10%), 29 (25%), 10 (9%), and 5 (4%) patients discontinued due to AEs in T12/PR24, T24/PR48, T24/P24, and PR48, respectively.
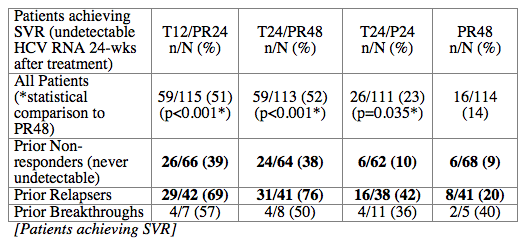
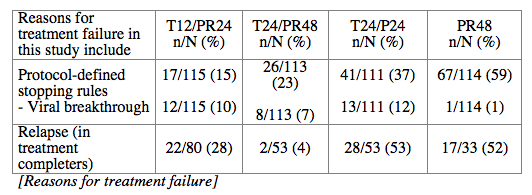
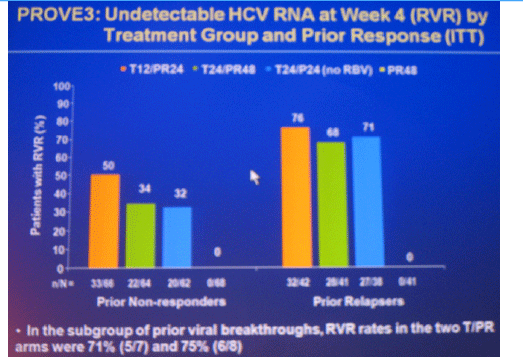
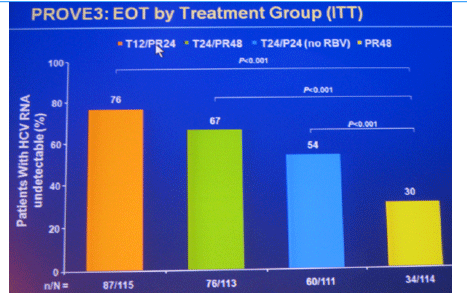
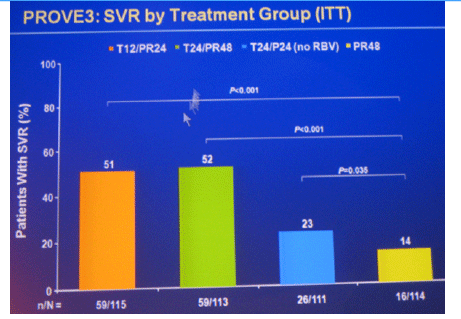
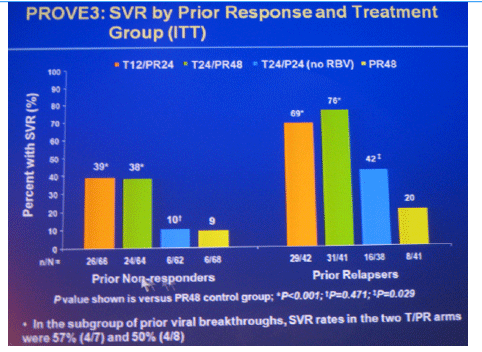
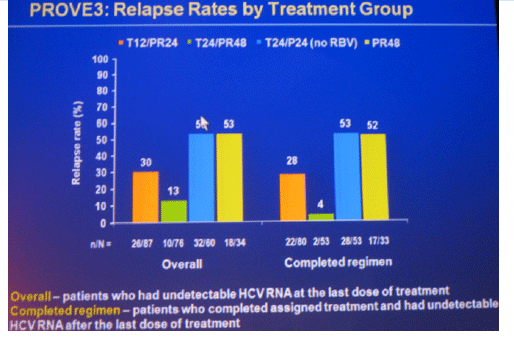
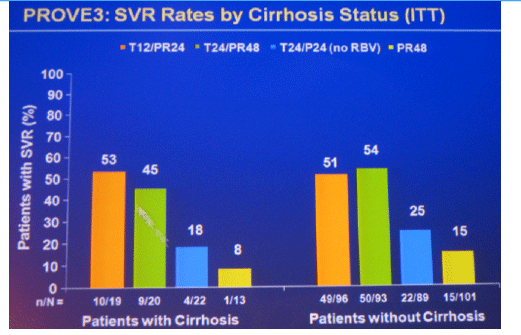
Conclusions: SVR rates in all treatment groups receiving T/PR regimens were significantly higher than with PR48. The general safety profile of T12/PR24 was similar to that observed in treatment-naïve patients. The higher relapse rate in T12/PR24 compared with T24/PR48 may warrant a total of 48-wks of PR in treatment-experienced patients. A Phase 3 study evaluating two T12/PR48 regimens is underway.




|
| |
|
|
|
|
|