 |
|
|
| |
Antiviral Activity of Filibuvir in Combination with
Pegylated Interferon Alfa-2a and Ribavirin for
28 Days in Treatment-Naive Patients Chronically
Infected with HCV Genotype 1
|
| |
| |
EASL April 23-26 2009 Copenhagen Denmark
Reported by Jules Levin
Antiviral Activity of the HCV Polymerase Inhibitor PF-00868554 ...
(AASLD) Oct 31-Nov 1 2008 ... Michael F. DeBruin1 1Infectious Diseases, Pfizer Global Research and Development, .... Poster 1898 presented at AASLD 2008.
www.natap.org/2008/AASLD/AASLD_18.htm
I. Jacobson,1 P.J. Pockros,2 J. Lalezari,3 E. Lawitz,4 M. Rodriguez-Torres,5 E. DeJesus,6 F. Haas,7 C. Martorell,8 R. Pruitt,9 K. Durham,10 S. Srinivasan,10 M. Rosario,10 S. Jagannatha,10 J. Hammond10
1Weill Cornell Medical College, New York, NY, USA; 2The Scripps Clinic, La Jolla, CA, USA; 3Quest Clinical Research, San Francisco, CA, USA; 4Alamo Medical Research, San Antonio, TX, USA; 5Fundacion de Investigacion de Diego and Ponce School of Medicine, Santurce, PR, USA; 6Orlando Immunology Center, Orlando, FL, USA;
7University of Oklahoma-Schusterman Clinic, Tulsa, OK, USA; 8The Research Institute, Springfield, MA, USA; 9Nashville Medical Research Institute and Maria Nathanson Center of Excellence at Saint Thomas Hospital, Nashville, TN, USA; 10Pfizer Global Research and Development, New London, CT, USA
AUTHOR CONCLUSIONS

Abstract
BACKGROUND: Filibuvir (FBV; formerly PF 00868554) is a non-nucleoside inhibitor
of HCV polymerase. As monotherapy (100-450 mg BID or 300 mg TID x 8 days), FBV
demonstrated dose-dependent inhibition of viral replication, with mean maximum
reductions in HCV RNA ranging from -0.97 to -2.13 (log10 IU/mL). In this study, the
safety and efficacy of FBV in combination with pegylated interferon alfa-2a (pegIFN) and
ribavirin (RBV) was evaluated.
METHODS: Eligible patients (treatment-naive, HCV genotype 1) were randomized 1:1:1:1
to receive FBV (200, 300 or 500 mg BID) or placebo in combination with pegIFN (180 ug/wk)
and RBV (1000/1200 mg/day) for 4 weeks. Patients are continuing on open label pegIFN/
RBV for an additional 44 weeks. Results from the Week 4 analysis are described here.
HCV RNA was measured using the Roche COBAS Taqman (LLD = 25 IU/mL).
RESULTS:
A total of 35 patients were enrolled and 34 completed Week 4. The majority
were Caucasian (69%), genotype 1a (80%) and male (51%). The most frequently reported
AEs were headache, fatigue, insomnia and nausea. There were no trends towards
increased frequency or severity of AEs with increasing dose of FBV. The incidence and
severity of laboratory test abnormalities was similar across treatment groups. There was
one treatment-related SAE (elevated creatinine) in the FBV 300 mg group, which resolved
following IV hydration. No other renal AEs were reported.
The mean reduction (log10 IU/mL) in HCV RNA at Day 4 for placebo (n = 8), 200 (n = 10), 300 (n = 9) and 500 (n = 8) mg BID was -0.58, -2.29, -2.72 and -2.83, respectively, and -2.10, -4.46, -4.67 and -3.62 at Day 28.
The percent of patients achieving RVR (undetectable HCV RNA by Week 4) for placebo, 200, 300 and 500 mg BID was 0%, 60%, 75% and 63%, respectively. Of those patients treated with 200, 300 and 500 mg BID who achieved a RVR, 33%, 33% and 80% achieved undetectable HCV RNA within two weeks of treatment, respectively.
CONCLUSIONS: The addition of FBV to pegIFN/RBV was well tolerated and markedly
increased the percentage of patients achieving RVR. Assessment of response to pegIFN/
RBV therapy subsequent to Week 4 is ongoing.
Background
Filibuvir (FBV; formerly PF-00868554) is a potent and selective non-nucleoside inhibitor of the hepatitis C virus (HCV) polymerase enzyme with a mean IC50 of 0.015 µM against genotype 1 polymerase.
FBV binds in the 'Thumb 2' site of the HCV polymerase.
FBV has potent in vitro antiviral activity with an overall mean EC50 against genotype 1 replicons of 0.059 µM. In vitro data indicate equipotent activity against genotypes 1a and 1b.
The safety, tolerability, pharmacokinetics, and antiviral activity of FBV monotherapy were investigated in healthy volunteers1 and treatment-naive HCV-infected patients2 in dose ranging studies. FBV elicited -0.97 to -2.13 (log10 IU/mL) mean maximum reductions in HCV RNA levels over eight days at doses of 100-450 mg BID, and 300 mg TID in patients infected with HCV genotype 1.
Sequence analysis of the NS5B region of isolates from patients participating in
FBV monotherapy studies demonstrated that substitutions at position 423 of the
NS5B polymerase were the predominant amino acid changes, arising in 11 of 24 FBV-dosed patients (46%) including 11 of 18 patients (61%) receiving doses of >100 mg BID.3
Objectives
To assess the antiviral activity, safety and tolerability of FBV (200-500 mg BID)
when administered in combination with pegylated interferon alfa-2a (pegIFN)
and ribavirin (RBV) over 28 days in treatment-naive patients infected with HCV
genotype 1.


Results
Patient demographics
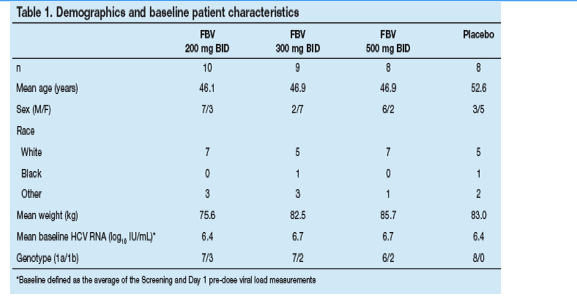
Demographic parameters and baseline patient characteristics in each treatment group are listed in Table 1.
Patient disposition
34 of the 35 patients randomized completed four weeks of therapy
- One patient in the FBV 300 mg BID treatment group withdrew consent on Day 3 owing to moderate nausea, vomiting and chills. Data for this patient are included in all safety analyses, but are only included in efficacy analyses where indicated.
PegIFN/RBV dose reductions
-- Three patients had either pegIFN (1 patient in FBV 200mg BID group) or RBV (1 patient in FBV 200 mg BID group and 1 patient in the 300 mg BID group) doses reduced during the first four weeks of the study due to anemia or neutropenia.
Viral response
All doses of FBV resulted in greater reductions in HCV RNA concentrations when added to a regimen of pegIFN/RBV than pegIFN/RBV alone (Table 2 and Figure 2).
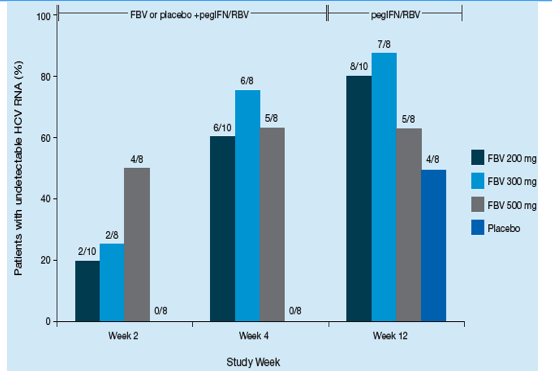
All doses of FBV significantly increased the proportion of patients achieving undetectable HCV RNA at Week 4 when added to a regimen of pegIFN/RBV compared with pegIFN/RBV alone (p<0.05). By Week 2, 0%, 20%, 25% and 50% of patients receiving placebo, FBV 200 mg, 300 mg or 500 mg BID had undetectable HCV RNA, respectively, and by Week 4, the respective values were 0%, 60%, 75% and 63%,
(Figure 3).
At Week 12, 50%, 80%, 88% and 63% of patients receiving placebo, FBV 200 mg, 300 mg or 500 mg BID had undetectable HCV RNA, respectively. All patients with undetectable HCV RNA at Week 4 remained undetectable at Week 12 (Figure 3).
Across all FBV dosing groups, 14 of 19 genotype 1a patients and 3 of 7 genotype 1b patients had a rapid virological response (RVR). Given the small number of genotype 1b patients in the study, it is unclear if there is a true difference in response between genotype 1a and 1b patients.

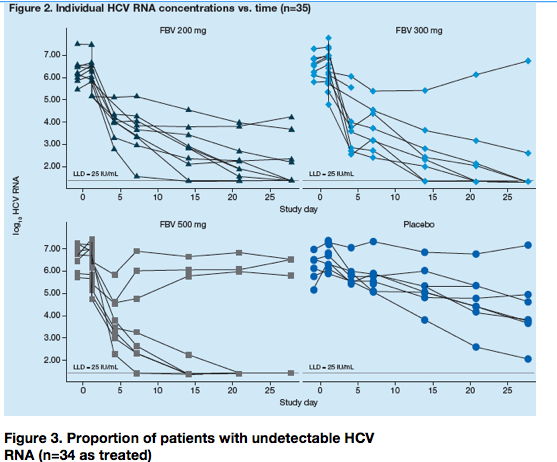
Virologic breakthrough
Virologic breakthrough (defined as a >0.5 log10 increase in HCV RNA from nadir) was observed in 1/10, 1/8 and 3/8 patients in the FBV 200 mg, 300 mg and 500 mg BID groups, respectively. All patients who experienced virologic breakthrough subsequently demonstrated a <2.0 log10 reduction in HCV RNA at Week 12, indicating a poor intrinsic response to pegIFN and RBV. This poor intrinsic response to pegIFN and RBV likely contributed to virologic breakthrough in these patients.
Six of the 34 patients (17.6%) completing 12 weeks of therapy demonstrated a
<2.0 log10 reduction in HCV RNA at Week 12. However, a disproportionate number of these patients (3 of 6) were randomized to the FBV 500 mg BID group (one patient with a <2.0 log10 reduction at Week 12 was in each of the other three treatment groups. The high rate of virologic breakthrough in the FBV 500 mg BID group was likely due to the higher number of patients with intrinsically poor response to pegIFN and RBV assigned by chance to this dosing group.
If those patients who subsequently demonstrated a <2.0 log10 reduction in HCV RNA at Week 12 are excluded from the analysis, 0/7 (0%), 6/9 (67%), 6/7 (86%) and 5/5 (100%) patients in the placebo, FBV 200 mg, 300 mg and 500 mg BID groups, respectively, achieved a RVR.
NS5B sequence analyses for those patients who demonstrated virologic breakthrough is currently ongoing.
Safety and tolerability
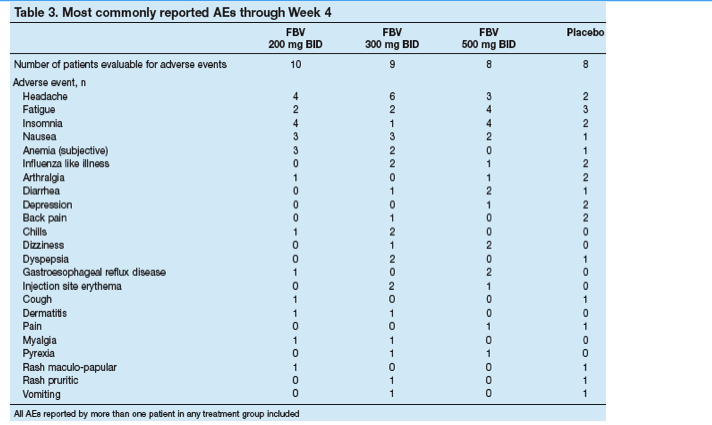
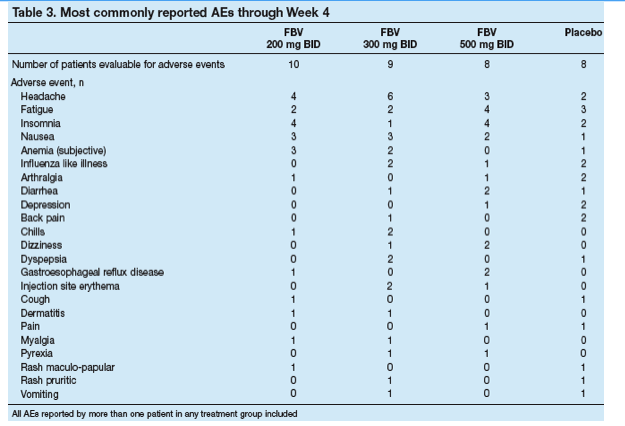
The most frequently reported AEs were headache, fatigue, insomnia and nausea (Table 3). There were no trends towards increased frequency or severity of AEs with increasing dose of FBV.
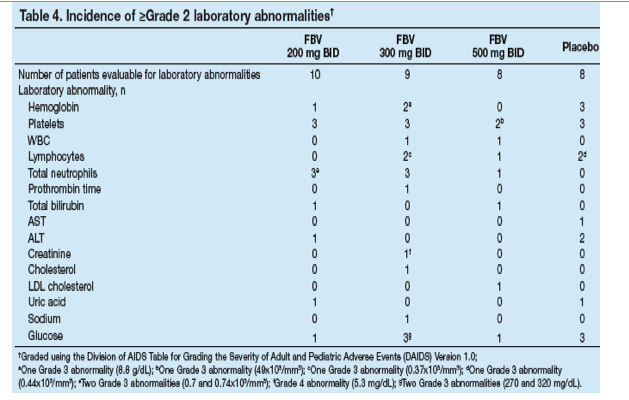
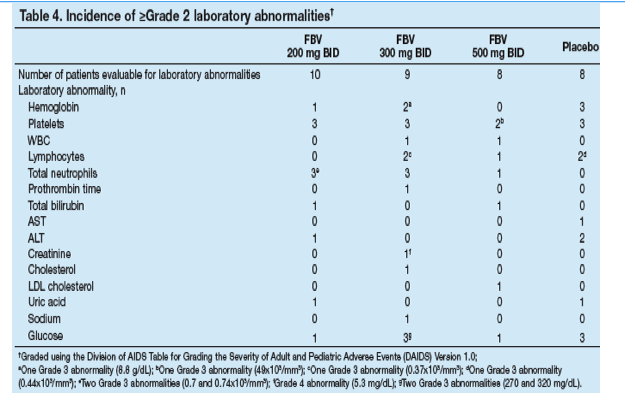
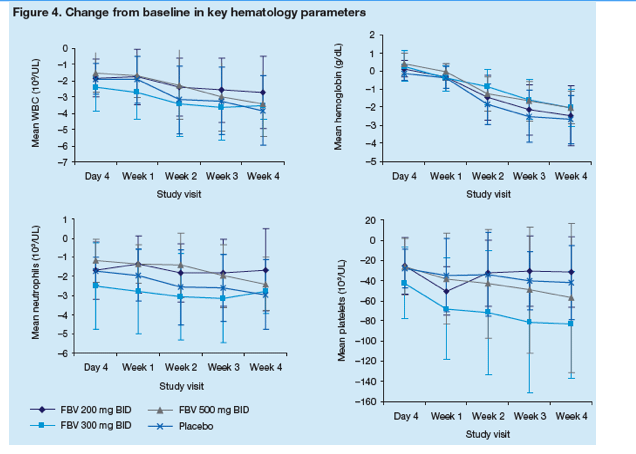
The incidence and severity of laboratory test abnormalities and the change from baseline in key hematology parameters were similar across treatment groups (Table 4, Figure 4).
One treatment-related serious AE (elevated creatinine) was reported in the FBV 300 mg BID group. The event occurred in a 58-year-old patient with a history of diabetes. The patient experienced nausea, vomiting and chills two days following initiation of study therapy. The patient discontinued all study medications on Day 3, and laboratory results
obtained at the early termination visit (Day 6) showed a serum creatinine of 5.4 mg/dL, BUN of 90 mg/dL and glucose of 320 mg/dL. Urine sediment and a renal ultrasound were unremarkable. Serum creatinine normalized with aggressive IV hydration. No other clinically significant creatinine elevations have been reported to date.
References
1. Hammond JL, et al. Safety, tolerability and pharmacokinetics of the HCV polymerase inhibitor PF-00868554 following multiple dose administration in healthy volunteers. Poster 1898 presented at AASLD 2008.
2. Hammond JL, et al. Antiviral activity of the HCV polymerase inhibitor PF-00868554 administered as monotherapy in HCV Genotype 1 infected patients. Poster LB11 presented at AASLD 2008.
3. Troke PJF, et al. Genotypic characterization of HCV NS5B following 8-day monotherapy with the polymerase inhibitor filibuvir (PF-00868554) in HCV-infected subjects. Poster 1795 presented at EASL 2009.
Disclosures
This study was sponsored by Pfizer Inc.
|
| |
|
|
|
|
|