 |
|
|
| |
TBR-652, A CHEMOKINE RECEPTOR 5 (CCR5) ANTAGONIST, DEMONSTRATES GOOD ORAL BIOAVAILABILITY AND DESIRABLE PHARMACOKINETIC (PK) AND SAFETY PROFILES IN HEALTHY VOLUNTEERS
|
| |
| |
Reported by Jules Levin
ICAAC Sept 2009 SF, CA
S. Palleja1, L. Wang-Smith2, R. Ogden1, D. Martin1, R. Driz1, J. Sapirstein1, 1Tobira Pharmaceuticals, Inc., Princeton, NJ; 2INDAPharma, LLC, Chapel Hill, NC
BACKGROUND
TBR-652, formerly known as TAK-652, is a promising CCR5 antagonist in phase 2 clinical development. A phase 1 single oral dose study showed rapid absorption and good bioavailability for TBR-652 tablets.1 Another study found that, although TBR-652 plasma concentrations were above the in vitro effective concentration level of 90% (EC90) in fasted subjects, a high-fat meal significantly increased the extent of TBR-652 bioavailability and reduced the intersubject variability in plasma TBR-652 concentrations.2 This healthy volunteer study (Tobira Therapeutics Study 652-1-102, Part II) investigated the safety, tolerability, and PK of multiple doses of TBR-652 administered once daily in one of two tablet formulations at doses ranging from 25 to 200 mg following a high-fat.
OBJECTIVES
To determine the safety and tolerability of multiple oral doses of TBR-652 in healthy adult subjects when administered once daily for 10 days.
To determine the pharmacokinetics of TBR-652 on Day 1 and Day 10 after once daily administration of various doses of TBR-652 for 10 days.
AUTHOR SUMMARY
TBR-652 was well tolerated at once-daily doses of up to 200 mg for 10 days.
TBR-652 was readily and well absorbed following single or repeated doses of the tablet formulations with a high-fat meal, achieving mean peak plasma concentrations in 4-6 hours.
The elimination kinetics of TBR-652 were similar across the dose range of 25 to 200 mg/day with a mean t1/2 of approximately 40 hours.
Plasma concentrations of TBR-652 achieved steady-state after 8 days of once daily dosing.
The mean accumulation ratios (Day 10 / Day 1) for AUC0-24 and Cmax of TBR-652 following repeated once-daily doses were 1.3-1.8 and 0.97-1.4, respectively, across the 25 to 200 mg/day dose range, regardless of formulation. These values were less than the predicted value of 2.9 based on a mean t1/2 of 40 hours and oncedaily dosing frequency, which might be attributable to slightly reduced bioavailability of TBR-652 after repeated doses.
A 25 mg/day F1 or F2 tablet achieved plasma exposures greater than the target therapeutic level of 2 ng/mL projected from in vitro studies of TBR-652.
There were no deaths or serious adverse events during the study.
Treatment-emergent adverse events considered possibly, probably, or definitely related to study drug reported by more than 1 subject in any treatment included constipation and headache.
Treatment-emergent adverse events were generally mild.
Two instances of elevated liver enzymes resolved off treatment and were most likely attributable to other underlying conditions not related to study drug (non-alcoholic fatty liver and muscle injury).
AUTHOR CONCLUSIONS
The safety and PK profiles of TBR-652 support further clinical development of TBR-652 in HIV-infected patients. A Proof of Concept study of TBR-652 in HIV infected patients is underway.
METHODS
Study Design: This was a double-blind, placebo-controlled, randomized, multiple-dose study. Five dose groups of 12 subjects each received TBR-652 in one of 2 tablet formulations daily for 5 days. Dosages ranged from 25 mg/day to 200 mg/day (Table 1). Within each dose group, subjects were randomly assigned in a ratio of 9:3 to TBR-652:placebo administered as either the previously used formulation (F1) or a new formulation (F2). All doses were taken with approximately 240 mL of water within 30 minutes after the start and within 10 minutes after the completion of a standard high-fat breakfast (approximately 150, 250, and 500-600 calories from protein, carbohydrate, and fat, respectively).
Subjects were sequestered at the clinical research unit (CRU) from the evening before the first dose until after the PK draw 48 hours after the last dose. Subjects returned to the CRU for PK draws at 60 hours, 72 hours, 96 hours, and 120 hours after the last dose. A final follow-up safety examination occurred on Day 20.
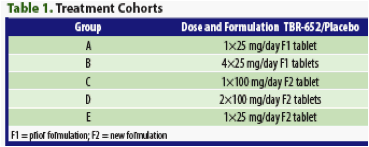
Study Population: Subjects were healthy males and females between 19 and 55 years of age who were willing to take appropriate precautions to prevent pregnancy and had no significant metabolic, hepatic, renal, hematologic, pulmonary, cardiovascular, or gastrointestinal (GI) disorders. Subjects had a body mass index (BMI) of 18 to 30 kg/m2 (inclusive), had no recent history (within 30 days) of clinically significant infection, and were negative serologically for hepatitis B and C and HIV. The use of concomitant medications or treatments was restricted except as needed for the subject's well-being.
Analyses: PK parameters of TBR-652 were calculated using noncompartmental methods and summarized by treatment/dose level using descriptive statistics. Analysis of variance (ANOVA) was used to determine dose proportionality between dose levels within the same tablet formulation. The accumulation ratios in Cmax and AUC0-24 were determined with ANOVA using a mixed-effect model. Safety and tolerability were assessed by monitoring adverse events, measuring vital signs, obtaining electrocardiograms (ECGs), and collecting clinical laboratory values from blood and urine samples.
RESULTS
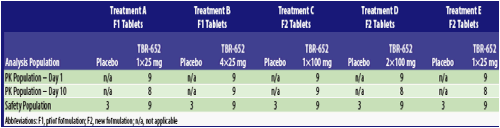
Demographics/Disposition: A total of 60 subjects enrolled in the study (Table 2). Three subjects receiving TBR-652 prematurely discontinued the study before the last dose on Day 10 (Table 3).
Table 2. Subject Demographics
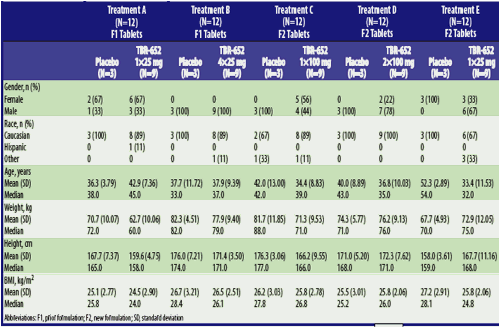
Abbreviations: F1, prior formulation; F2, new formulation; SD, standard deviation
Table 3. Analysis Populations
Pharmacokinetics
Figure 1. Mean Plasma Concentration vs. Time Curves of TBR-652
in 2 Tablet Formulations, Day 1, Linear and Semilog Plots
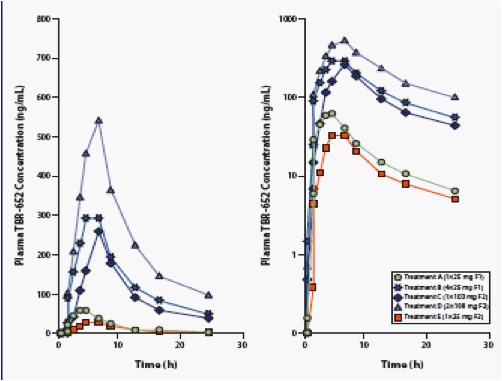
Figure 2. Mean Plasma Concentration vs. Time Curves of TBR-652
in 2 Tablet Formulations, Day 10, Linear and Semilog Plots
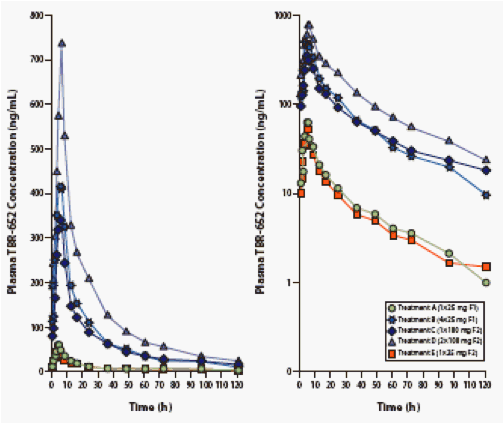
Table 4. Summary of TBR-652 Pharmacokinetic Parameter Estimates by Dose and Day
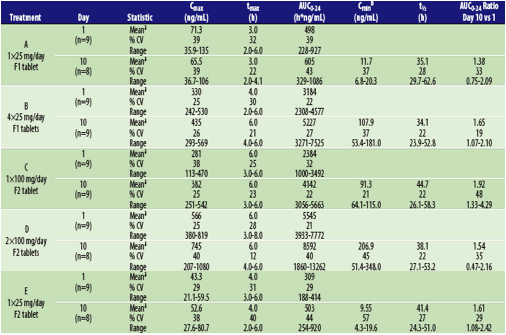
Abbreviations: CV, coefficient of variation; F1, prior formulation; F2, new formulation. aValues are medians for tmax and t1/2. bMean Cmin was average pre-dose concentrations on Days 9, 10 and 11.
Figure 3. Individual and Mean Day 10 Cmax for All Treatments
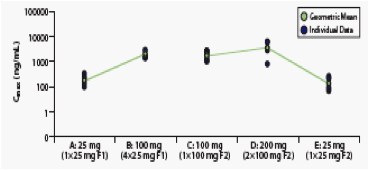
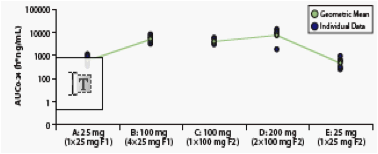
Figure 4. Individual and Mean Day 10 AUC0-24 for All Treatments
Safety and Tolerability: TBR-652 was generally well-tolerated when administered as 25 to 200 mg once daily over 10 days to healthy volunteers. There were no deaths or serious adverse events (SAEs) during the study (Table 5). One treatment-emergent adverse event was classified by the investigator as grade 3/4, 3 were classified as grade 2, and all others were classified as grade 1 (mild). One subject each in Treatments A and E
(the lowest doses studied) experienced adverse events that resulted in treatment discontinuation. The first subject experienced asymptomatic grade 4 aspartate aminotransferase (AST) and alanine aminotransferase (ALT) elevations on Day 7, at which time study drug was discontinued. The subject reported no AEs, no physical signs were noted by the Investigator, and liver function tests returned to normal by Day 26. The event was attributed to pre-existing, unreported non-alcoholic fatty liver disease. The other subject had a grade 2 AST elevation and an elevated lactate dehydrogenase (LDH) on Day 8, which resolved completely and were thought to be caused by a mild muscle injury. Neither event met Hy's law, a prognostic indicator of severe liver injury. No notable effects were noted on vital signs or ECGs in any subject.
Table 5. Treatment-Emergent Adverse Event Reporting Profile (Safety Population)
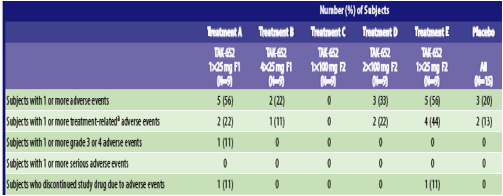
Treatment-emergent adverse events reported by more than 1 subject in any treatment are noted in Table 6. Treatment-related adverse events reported by more than 1 subject in any treatment included constipation: 2 (22%) subjects each in Treatments D and E, and headache: 2 (13%) subjects in the placebo group.
Table 6. Summary of Treatment - Emergent Adverse Events (Safety Population)
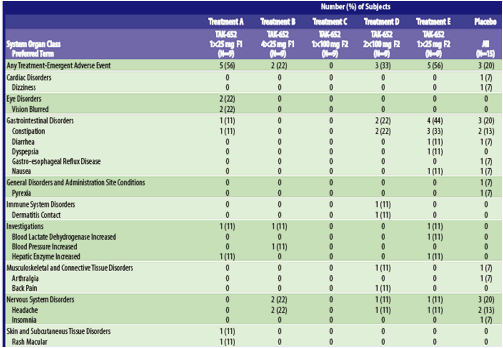
Abbreviations: F1, prior formulation; F2 new formulation
REFERENCES
1. Palleja S, et al. Pharmacokinetics (PK), Safety, and Tolerability of Single Doses of the Chemokine C-C Receptor 5 (CCR5) Antagonist TBR-652 in Healthy Volunteers. 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention. Cape Town, SA. July 19-22, 2009. Poster #WEPEB251.
2. Palleja S, et al. Effect of Food on the Relative Bioavailability and Pharmacokinetics of TBR-652, a Potent New Chemokine C-C Receptor 5 (CCR5) Antagonist. 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention. Cape Town, SA. July 19-22, 2009. Poster #WEPEB252.
|
| |
|
|
|
|
|