 |
|
|
| |
Clinical Pharmacology at CROI 2010: Advances in Antiretroviral Discovery, Drug Interactions, Compartmental Penetration, and Adverse Reactions
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
986000 Nebraska Medical Center
Omaha, NE 68198
The 17th Conference on Retroviruses and Opportunistic Infections (CROI) was held in San Francisco, CA from February 16-19, 2010. CROI continues to be the premier HIV-focused scientific meeting. The 17th CROI had many abstracts related to clinical pharmacology. In this report I will highlight those I think are of broad interest or might benefit from some expert clarification. I will discuss abstracts in four broad categories: advances in antiretroviral discovery, drug interactions, compartmental penetration and adverse reactions.
Abbreviations
%CV, percent coefficient of variation
ABC, abacavir
ACTG, adult AIDS clinical trials group
APV, amprenavir
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
ATV, atazanavir
Cmin, minimum drug concentration
CNS, central nervous system
CSF, cerebrospinal fluid
CYP, cytochrome P450 drug metabolizing enzymes
DRV, darunavir
ddI, didanosine
EFV, efavirenz
EVG, elvitegravir
FTC, emtricitabine
ETR, etravirine
fAPV, fosamprenavir
IC50, concentration of drug required to inhibit viral replication in vitro by
50%
IDV, indinavir
IM, intramuscular
IQ, inhibitory quotient
3TC, lamivudine
LPV, lopinavir
MVC, maraviroc
NVP, nevirapine
NRTI, nucleoside reverse transcriptase inhibitor
NNRTI, non-nucleoside reverse transcriptase inhibitor
PACTG, pediatric AIDS clinical trials group
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamic
PG, pharmacogenetics/pharmacogenomics
PK, pharmacokinetic
PI, inhibitor of HIV protease
r or RTV, ritonavir
RAL, raltegravir
SQV, saquinavir
SC, subcutaneous
TDM, therapeutic drug monitoring
TDF, tenofovir
TPV, tipranavir
ZDV, zidovudine
I. Advances in Antiretroviral Discovery
Abstract #58LB reported the results of 2 small phase II studies in antiretroviral-naïve persons of the investigational integrase inhibitor elvitegravir (EVG) and the Gilead booster, GS-9350 (now given the generic name cobicistat). Recall that GS-9350 was first reported at the 2009 CROI (Abstract #40) as an agent that was a potent inhibitor of CYP with no antiviral activity against HIV and reduced in vitro effects on adipocytes. One study compared the once daily "QUAD" tablet of EVG, GS-9350, TDF and FTC, with EFV/TDF/FTC (Atripla). The second compared the ability of GS-9350 (150 mg) to boost ATV vs RTV, when also given with TDF/FTC. The EVG, GS-9360/TDF/FTC regimen produced a virologic effect (% with HIV RNA < 50 copies/mL) that was noninferior to EFV/TDF/FTC. The ATV regimen boosted with GS-9350 also achieved a noninferior virologic response to ATV boosted with RTV. These studies are both important steps forward in the development of a new integrase inhibitor (EVG) and a new pharmacokinetic booster, GS-9350. However, as is always the case in drug development, there are surprises and every pharmacologic agent has some undesired, drug related side effects. The table below shows the adverse side effects reported during this presentation of these studies.
Event QUAD
N=48 EFV/TDF/FTC
N=23 GS-9350
(N=50) RTV
(N=29)
Grade 3 or 4 adverse events 0 2 (9%) 2 (4%) 0
Adverse events leading to discontinuation of study drugs 0 1 (4%) 2 (4%) 0
Elevation of total cholesterol (grade 3-4) 4 (9%) 2 (10%) 3 (6%) 0
Elevation of serum creatinine
(grade 1) 1 (2%) 0 6 (12%) 0
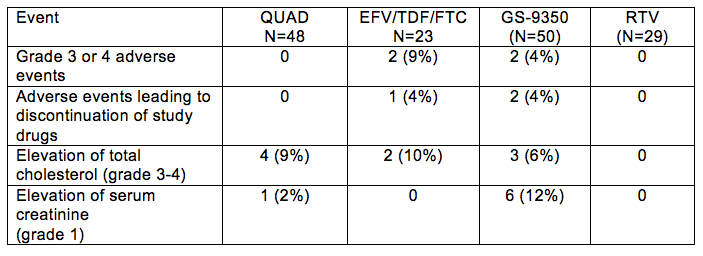
Here is my take on what these mean, with the caveat that we are dealing with early knowledge from studies in very small numbers of persons. First, the accumulating safety and virologic response data for the investigational integrase inhibitor EVG, now combined in a single tablet with GS-9350, TDF and FTC, continues to be positive. Second, GS-9350 seems to have CYP inhibiting properties approximately equivalent (on a mg per mg basis) to RTV. This evidence comes from the virologic noninferior response of ATV+GS-9350 vs. ATV+RTV, and also EVG/GS-9350/TDF/FTC vs. EFV/TDF/FTC. Third, GS-9350 is not the pure CYP inhibitor we had hoped for. The elevations in total cholesterol seen in both the EVG/GS-9350/TDF/FTC regimen and ATV/GS-9350/TDF/FTC regimen provide evidence that GS-9350 has an adverse effect on lipids. Fourth, GS-9350 has an adverse effect of elevating serum creatinine. Evidence was shown that this effect was an inhibition of the kidney tubular secretion of creatinine and not an actual decrease in measured creatinine clearance. However, more needs to be learned about this effect and whether there is indeed no pathology associated with it. The higher rate of serum creatinine elevations when GS-9350 was given with ATV vs. EVG is interesting, and needs further investigation in my opinion. Finally, the approach to the clinical management of this adverse effect needs careful development and validation. An elevation in serum creatinine usually needs clinical investigation because it may indicate impaired renal function or nephrotoxicity, especially in patients who are taking drugs known to cause kidney toxicity (such as TDF). Thus, the challenge is going to be how to differentiate between a "harmless" elevation in serum creatinine vs. nephrotoxicity.
Abstract 49 was a superb presentation from investigators at Katholieke University in Belgium of a rational drug design approach they employed to develop small molecule inhibitors of the LEDGF and HIV integrase interaction. They demonstrated HIV inhibition of micromolar concentrations and the lack of cross-resistance with RAL and EVG. I am eagerly awaiting news that these compounds can be moved from the lab to the clinic. Abstract 55 reported information on the drug design approach to the Shionogi/GSK integrase inhibitor S/GSK1349572. Phase 2 studies of this agent have demonstrated a 2.5 log10 reduction in HIV-RNA with a 50 mg once daily dose. Abstract 53 and 598 presented information on TBR 652, a CCR5 antagonist. Doses between 25 to 150 mg once daily have been given for 10 days. The steady-state concentrations of TBR 652 were strongly related to the drop in HIV-RNA, and up to a 1.8 log10 reduction in HIV-RNA has been observed. Short-term tolerance was acceptable. Further Phase 2 studies of this agent are planned, and seem warranted. These select examples of new drug discovery and development indicate the promise of future options for treatment of HIV infection.
II. Drug Interactions
CROI has always provided an important venue for information on new drug-drug interactions, and this 17th conference was no exception. Here are some that I found to be the most surprising, interesting and clinically important.
In the category of surprising, Abstract 603 reported an increase in the concentrations of the antimalarial agent lumefantrine when given with NVP in HIV-infected persons. In this study of 36 persons, the median lumefantrine day 7 concentrations were 632 ng/mL in the NVP treatment group and 344 ng/mL in the ART-naïve (no NVP) arm (p<0.0001). Lumefantrine is metabolized via CYP and NVP is regarded as a potent inducer of CYP. Thus, what we would have expected is for lumefantrine concentrations to be reduced when given with NVP. The day 7 plasma concentration of lumefantrine has been shown to be a useful marker of the anti-malarial effect. When the day 7 plasma lumefantrine concentration was below 280 ng/mL, 49% of uncomplicated malaria patients failed artemether-lumefantrine treatment. So now to make this study more interesting, one-third (6/18) of the ART-naïve persons had day 7 lumefantrine concentrations below this cutoff value of 280 ng/mL, compared with 6% (1/18) of those taking NVP-containing ART. Thus, not only was this interaction surprising, but it appeared to be therapeutically beneficial! Malaria and HIV combined account for 4 million deaths annually. The combination of artemether-lumefantrine is a recommended first-line treatment for malaria. NVP is widely used in Africa. The frequency with which the combination of lumefantrine and NVP may be coadministered is high. The high rate of subtherapeutic lumefantrine day 7 concentrations in the HIV-infected persons who were not taking NVP needs further investigation. The mechanism of the interaction between lumefantrine and NVP needs to be identified. And, further investigations to clarify clinical significance and management strategy for the combination of lumefantrine and NVP, but generally for the concomitant administration of antimalarial and antiretroviral drugs are urgently necessary.
The TRIO study has recently been published (Clinical Infectious Diseases 2009;49:1441-9) providing information that the combination of RAL, ETR and DRV/r in patients with multidrug-resistant virus was well tolerated and produced an overall 86% proportion of subjects with HIV-RNA < 50 copies/mL at week 48. Pharmacokinetic information on this combination has been needed because drug interactions studies have shown that ETR concentrations are decreased when given with DRV/r and are slightly increased with given with RAL; DRV concentrations are increased by ETR; and RAL concentrations are decreased when given with ETR. Abstract 606 provided PK data in HIV-infected persons for RAL, DRV and ETR receiving this combination in the TRIO study. DRV concentrations were slightly, approximately 15% increased when given with RAL and ETR. This is consistent with data in healthy volunteers indicating ETR increases DRV concentrations. The AUC of RAL was also slightly increased, approximately 30%, with the combination of ETR and DRV. Neither the increase in DRV or RAL concentrations would appear to be clinically important, and this would be supported by the TRIO study indicating this triple regimen was well tolerated. While the DRV and RAL concentrations in this PK study could be compared before and after the addition of ETR (because of the design of TRIO), the concentrations of ETR had to be compared with historical controls. When compared with those in HIV-infected persons receiving 200 mg twice daily with DRV, the ETR AUC is reduced by approximately 45% and the Cmin is reduced by 38%. The resulting concentrations approximate those with ETR is given with tipranavir/ritonavir, which is not a recommended combination because of the reduction seen in ETR concentrations. Though the TRIO clinical data provide some assurance of the virologic potency of the ETR/RAL/DRV/r regimen, it does not provide complete confidence that this apparent reduction in ETR concentrations has no virologic consequence. I'd advise clinicians using this regimen to monitor the response of patients closely, and suggest some additional pharmacology investigations into this combination, particularly ETR concentrations.
Abstract 934 evaluated the effect of EFV on the pharmacokinetics of levonorgestrel (LNG). LNG, known as Plan B, is used for prevention of pregnancy following unprotected intercourse or suspected contraceptive failure. Plan B is administered as soon as possible, but within 72 hours after intercourse. The use of LNG for emergency prevention of pregnancy has been shown to reduce pregnancy rates by 89%. In this study in healthy women, the PK of LNG were evaluated following a single dose of 0.75 mg before and after 14 days of EFV, 600 mg once daily. The AUC of LNG was reduced significantly by 58% and the elimination half-life decreased from 10.3 hours to 5.5 hours. The minimum effective dose of Plan B (LNG) is not known, however, these data raise concern as to whether the 0.75 mg dose will retain effectiveness if given to women who are also receiving EFV. These data indicate that additional studies of higher LNG doses and alternative dosing strategies are needed, and that the use of dual methods, including a barrier device, of contraception in women receiving EFV need continued reinforcement.
III. Compartmental Penetration
Considerable attention was devoted to the topic of compartmental penetration of ARVs. Dr. Deanna Kroetz discussed this topic and the role of membrane transporters in the pharmacology plenary, session 34, and interested readers who didn't hear her excellent talk might wish to watch the webcast. Several abstracts were presented on the penetration of ARVs into the genital tract, and there was a themed poster discussion (session 44). The bullet point findings from the genital tract penetration abstracts follow below.
DRV In 47 HIV-infected men receiving DRV/RTV, 600/100 mg twice daily, the DRV semen to blood plasma penetration averaged 8.6%. Semen penetration approximated the fraction of DRV that was free, or not bound to proteins in the plasma. Over the course of this study, among 45 subjects, 6 had detectable levels of HIV-RNA in semen (> 200 copies/mL) with corresponding undetectable levels of HIV-RNA in blood plasma. (Abstract 610)
In 18 HIV-infected men receiving DRV/RTV, 800/100 mg once daily, the DRV semen to blood plasma penetration averaged 17%. One subject had detectable levels of HIV-RNA in semen but undetectable HIV-RNA in blood. (Abstract 611)
MVC In 12 healthy men, the MVC concentrations in semen were about 40% lower than in plasma; MVC concentrations in rectal tissue were about 10-fold higher than in plasma. (Abstract 85)
In 12 HIV-infected men, MVC concentrations in semen were about 28% lower than concentrations in plasma. MVC was measured in cerebrospinal fluid (CSF) samples and the penetration averaged 2% of blood plasma concentrations. Actual CSF concentrations averaged 2.6 ng/mL and were within the broad range of the 50% inhibitory concentrations of MVC for HIV-1 or higher. (Abstract 612)
RAL In 14 HIV-infected women, RAL concentrations in cervicovaginal fluid were about 2.3-fold higher than in blood plasma. (Abstract 608)
In 8 healthy males, RAL concentrations in semen were from 1.6-fold higher than in blood plasma at 2-4 hours post dose to 6.5-fold higher at the end of the dosing interval (12 hours). (Abstract 609)
TDF/FTC In macaques, concentrations of TDF and FTC were demonstrated in lymphoid and rectal tissue. A single pre-exposure dose of TDF/FTC, however, did not protect against SHIV infection. A combined pre-exposure and post-exposure dose of TDF/FTC was highly protective. (Abstract 83)
These abstracts all add to our knowledge of the genital tract penetration of ARVs. What is interesting to me is the differences among penetration of drugs in the same class. For example, previous work has shown that among the NRTIs, penetration into cervicovaginal fluid can range from 5% for d4T to 400% for 3TC. There is also evidence for a gender difference in ARV penetration into the genital tract. MVC has been shown to have a penetration into cervicovaginal fluid approximately 3-fold higher than blood plasma. In men, however, the AUC of semen concentrations was 38% lower than the AUC in blood plasma. The data presented at this meeting on DRV makes a good point that penetration ratios or percents don't tell the whole story. While DRV penetration into semen averaged 10-20% of blood plasma concentrations, the actual concentrations of DRV in the semen were all above the protein binding corrected 50% inhibitory concentration of DRV for wild-type HIV-1 in Abstract 611 and above in the vast majority of subjects in Abstract 610. While most of the subjects in the 2 DRV studies had undetectable levels of HIV-RNA in semen, in those few who did have detectable HIV-RNA in semen they all had undetectable levels of HIV-RNA in blood plasma. This tells us that it is not a certainty that someone who has undetectable HIV-RNA in plasma will have undetectable HIV-RNA in the genital tract compartment. Clearly, we need to learn more about the determinants of ARV penetration into the genital tract of men and women, and the virologic consequences of drug in that compartment.
For those who would like an up-to-date slide of what is known about ARV penetration into the genital tract, I would refer you to Steve Taylor's poster presentation at this meeting, which has a summary slide of what was known and with all of the CROI 2010 information added (#611). Any users of this slide, please make sure you acknowledge Steve for his efforts to rapidly put all of this information together. Go to this link, and enter abstract # 611: http://www.retroconference.org/AbstractSearch/Default.aspx?Conf=19
IV. Special Considerations and Populations
Administration of combination EFV/TDF/FTC tablet (Atripla) is presently limited to individuals who can swallow tablets. Abstract 605 presented a PK evaluation of a liquid formulation of EFV/TDF/FTC pharmacist-compounded from the crushed tablet compared with the commercial fixed dose combination (FDC) tablet. 14 healthy volunteers received both formulations in a randomized manner. Concentrations were compared with the usual FDA bioequivalence assessment approach (90% confidence interval within a ratio of liquid to tablet of 0.8 to 1.25). For EFV, the Cmax ratio fell slightly below the desired ratio range while AUC was slightly above. For TDF, the Cmax and AUC were approximately 40% and 20%, respectively, higher with the liquid formulation. FTC Cmax and AUC fell within the standard bioequivalence ratio range. These data indicate that the liquid formulation does not meet the FDA standard bioequivalent definition for EFV and TDF. However, if a patient who was stabilized on the EFV/TDF/FTC tablet absolutely required administration of a liquid formulation for a short period of time, I think the risk/benefit of this approach would be acceptable.
In contrast, abstract 877 evaluated the PK of LPV/r tablets (200/50 mg) either as the whole tablet or crushed in children who had all been taking LPV/r for > 4 weeks. In this study of 12 children with a median age of 13 years, both LPV and RTV concentrations were significantly reduced when given as the crushed tablet. The crushed to whole tablet ratio of AUC for LPV was 0.60 and was 0.61 for RTV, indicating concentrations were 40% lower. These data would suggest that the LPV/r tablet formulation should not be crushed for administration, in either children or adults.
V. Adverse Drug Reactions - Complications of ARV Use
The issue of the use of antiretroviral agents increasing the risk of myocardial infarction (MI) received considerable attention this year as it has at the last two CROI conferences. In my mind, what has generated the most controversy has been the finding that the NRTIs ABC and ddI were associated with an increased risk of MI. An updated analysis from D:A:D was published in the February 1, 2010 edition of Journal of Infectious Diseases (2010;201:318-30) and was accompanied by an excellent editorial by Judy Aberg (2010;201:315-17). In this most recent analysis from D:A:D, with an additional year of follow-up, ABC and ddI remained the only NRTIs associated with a significantly increased risk of MI. Particularly to a pharmacologist, what has been missing to date is the mechanism by which ABC and ddI increase the risk of MI?
At CROI 2010 there was some progress towards identifying a potential mechanism(s) for ABC. There were 3 abstracts I thought were informative, #s 708, 716 and 717. Abstract 708 reports that HIV-infected persons had higher hsCRP, a measure of inflammation, than did uninfected controls. In the HIV-infected persons, a higher hsCRP was associated with impaired flow mediated dilation (FMD) of the brachial artery, which is a measure of endothelial function and is predictive of future cardiovascular events. Finally, ABC exposure was associated with impaired FMD. This work suggests an adverse effect of ABC on vascular function. The authors (Hsue P et al) of this abstract have published recent work in this area, see AIDS 2009;23:2021-27.
One hallmark of vascular disease is leukocyte (white blood cell) accumulation in the vessel wall. Abstract 716 explored the effect of ABC on human leukocytes and human endothelial cells. These investigators found that ABC increased leukocyte adhesion to endothelial cells in a concentration-related manner. Additionally, these investigators demonstrated the specific protein that was enhanced by ABC, and showed that blocking this protein prevented ABC-induced leukocyte adhesion.
Platelets play a critical role in acute MI, and ABC has been suggested to promote platelet aggregation. Abstract 717 evaluated whether ABC and its intracellular metabolite, carbovir-triphosphate, might inhibit guanylyl cyclase, which is a negative regulator of platelet function (i.e. less guanylyl cyclase increases platelet reactivity/aggregation). Indeed, carbovir-triphosphate was shown to inhibit guanylyl cyclase, and platelet activation was increased when platelets were incubated with ABC. This work now provides a potential mechanistic link between ABC exposure, increased platelet aggregation, and MI. Interesting, these authors commented that if this is the mechanism by which ABC increases the risk of MI that it might be mitigated by co-administration with aspirin (because of the anti platelet aggregation properties of aspirin).
Collectively, I thought these 3 abstracts advanced our knowledge of the mechanism. Why we need to understand this is really captured by the 3rd abstract. If we know why it happens then it provides information on whether we may be able to prevent this adverse reaction.
As true for most aspects of the treatment of HIV infection, more work to do - - stay tuned!
CROI 2010 Pcol-NATAP-031610.doc
|
| |
|
|
|
|
|