 |
|
|
| |
InterMune Reports Virologic Response of Ritonavir-Boosted Danoprevir (RG7227/ITMN-191) in Patients with Chronic Hepatitis C
|
| |
| |
-- Regimen Delivered Potent Viral Response and was Safe and Well-Tolerated --
BRISBANE, Calif., April 15 /PRNewswire-FirstCall/ -- InterMune, Inc. (Nasdaq: ITMN) today announced results of all three cohorts from a 15-day Phase 1b multiple-ascending-dose (MAD) study of low doses of danoprevir (also known as RG7227 and ITMN-191) boosted by low-dose ritonavir (RTV) in patients chronically infected with hepatitis C virus (HCV) genotype-1. The results announced today were reported in an oral presentation at the 45th Annual Meeting of the European Association for the Study of the Liver (EASL) in Vienna, Austria by Edward Gane, M.D., Associate Professor, University of Auckland, and Director, Auckland Clinical Studies Limited.
Dr. Gane commented, "The results from this study indicate that robust viral kinetics in treatment- naive patients can be achieved by ritonavir boosting of very low doses of danoprevir. We look forward to the results of the two 12-week cohorts in prior SOC null responders recently added to this study which will provide further insights into the antiviral efficacy and safety profile of low doses of ritonavir-boosted danoprevir."
Dan Welch, Chairman and CEO of InterMune commented, "Importantly, 18 of 25 patients (72%) treated with ritonavir-boosted danoprevir had HCV RNA levels below the limit of detection after only 15 days in this study. These results are at least as strong as those of the 14-day observation period of the 12 week Phase 2b study using un-boosted, high doses of danoprevir with exposure substantially higher than ritonavir-boosted doses. The potential impact of lower systemic exposure on the long-term safety profile of danoprevir will be further evaluated in future studies. The collective results to date appear to confirm that our objective with ritonavir-boosting is realistic."
Viral Kinetic and Pharmacokinetic Performance of Ritonavir-Boosted Danoprevir
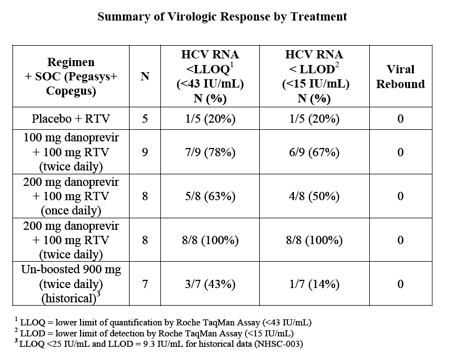
The Phase 1b study examined three dosage cohorts of danoprevir to date: 100 mg twice daily, 200 mg once daily, and 200 mg twice daily. Each was administered for 15 days in combination with 100 mg ritonavir on the same schedule, and with standard-dose PEGASYS and COPEGUS, the current standard of care (SOC).
The combination of danoprevir 100 mg administered twice daily with 100 mg twice-daily ritonavir resulted in 78% of patients achieving a level of HCV RNA below the lower limit of quantification (LLOQ; <43 IU/mL, 7 out of 9 patients total), and 67% of patients having HCV RNA levels below the lower limit of detection (LLOD; <15 IU/mL, 6 out of 9 patients total) by day 15.
A 200 mg once-daily dose of danoprevir with a 100 mg once-daily dose of ritonavir resulted in 63% of patients with HCV RNA levels below LLOQ (5 out of 8 patients total) and 50% of patients with HCV RNA levels below LLOD (4 patients out of 8 patients total) after only 15 days. At the highest daily dose, 200 mg twice-daily danoprevir in combination with 100 mg twice-daily ritonavir, 100% of patients achieved HCV RNA below LLOQ and LLOD (8 out of 8 patients total) after 15 days.
No patient experienced virologic rebound in any dosage group during the 15-day study.
The pharmacokinetic profile of ritonavir-boosted danoprevir was more favorable than that observed in previously reported studies conducted with much higher doses of un-boosted danoprevir. At 100 mg danoprevir dosed twice daily with 100 mg ritonavir, AUC and Cmax were approximately 16-fold and 23-fold lower, respectively, than that provided by 900 mg twice-daily un-boosted danoprevir (historical Phase 1 and 2 data).
Safety and Tolerability
The ritonavir-boosted combination showed a favorable safety and tolerability profile. The most commonly reported adverse events (AEs) were headache, nausea and diarrhea and these had a similar incidence to previously reported studies of danoprevir in combination with standard of care (SOC).
Amended Protocol - 12 Weeks of RTV-boosted Danoprevir
The companies reported that the Phase 1b MAD ritonavir-boosting study protocol has been amended to add two cohorts which will examine 12 weeks of low doses of ritonavir-boosted danoprevir plus SOC in prior SOC null responders. The doses to be examined in the expanded protocol are 100 mg twice daily and 200 mg once daily, each in combination with 100 mg ritonavir on the same schedule, plus the standard dose and regimen of SOC. The first patient in the amended protocol was enrolled in late March 2010.
About Ritonavir Boosting
Ritonavir boosting is an option to enhance and improve pharmacokinetic profiles of protease inhibitors. It is well established in the treatment of HIV where it leads to more convenient dosing, reduced resistance development and high efficacy. Not all HCV protease inhibitors are suitable for ritonavir boosting. InterMune announced on August 6, 2009, ritonavir boosting of danoprevir showed promise in a Phase 1 drug-drug interaction study in healthy volunteers. The results of this study demonstrated that the co-administration of low-dose ritonavir increased danoprevir concentration 12 hours post dose by approximately 27 times, with the effect on AUC being roughly 5 times and Cmax being roughly 3 times. These results guided the selection of the doses of danoprevir reported today in the Phase 1b MAD study in HCV patients.
About Danoprevir (RG7227/ITMN-191)
Danoprevir (also known as RG7227 and ITMN-191) is a potent, macrocyclic inhibitor of HCV NS3/4A protease activity being developed in collaboration with Roche. The safety and antiviral activity of danoprevir is expected to be further evaluated in a Phase 2b study of danoprevir in combination with low doses of ritonavir and the current standard of care, and is also under clinical investigation in combination with the NS5B nucleoside polymerase inhibitor RG7128 in the INFORM clinical development program.
InterMune (ITMN) Updates on Phase 2b of Danoprevir in Combination with PEGASYS and COPEGUS
April 14, 2010 7:09 AM EDT
InterMune, Inc. (NASDAQ: today announced top-line results from the planned Week 12 week interim analysis of the Phase 2b randomized, partially-blind study evaluating the hepatitis C virus (HCV) protease inhibitor danoprevir (also known as RG7227 and ITMN-191), administered for 12 weeks in combination with PEGASYS (pegylated interferon alfa-2a) and COPEGUS (ribavirin), compared with placebo for the same duration plus PEGASYS and COPEGUS.
Dan Welch, Chairman, Chied Executive Officer and President of InterMune, said, "The RVR and cEVR results reported today are among the very best reported by any DAA compound to date, reinforcing our view that danoprevir may potentially play a meaningful role in the treatment of HCV patients. With Roche, we are now focused on making the critical dose and regimen selection decision for future development plans for ritonavir-boosted danoprevir, an approach that appears to deliver strong efficacy and offer attractive advantages of dosing convenience and increased safety margin."
Frank Duff, M.D., Head of Roche's Clinical Development for Virology, said, "Future development of danoprevir is expected to be conducted in combination with ritonavir at total daily doses that are 10-25% of those examined in this study. The pharmacokinetics/pharmacodynamics and safety data from this large, well-controlled study, in addition to other data being collected, will be very helpful in our efforts to choose the optimal dose and regimen for the ritonavir-boosted danoprevir global development program, including the all-oral, direct-acting antiviral INFORM component of the program."
Safety and Tolerability
The analysis of the safety data is preliminary in nature and additional evaluation is ongoing. In the interim safety analysis, serious adverse events (SAEs) were generally balanced across all four treatment groups. The incidence of treatment-emergent Grade 4 (>10x ULN ALT elevations was 0%, 1%, 6% and 0% in the 300 mg three-times daily, 600 mg twice daily, 900 mg twice daily and placebo groups, respectively. In the danoprevir treatment groups, these elevations occurred generally between weeks 6-8 or later and were reversible after discontinuation of danoprevir. Dosing of the 900 mg arm was stopped based on this safety signal. Treatment-emergent Grade 3 or Grade 4 neutropenia was reported in 24%, 25%, 31% and 16% of patients in the 300 mg three-times daily, 600 mg twice daily, 900 mg twice daily and placebo groups, respectively. The incidence of rash and anemia was comparable across all treatment groups, including the placebo group (SOC + placebo).
|
| |
|
|
|
|
|