 |
|
|
| |
DIONE - 24 week efficacy, safety, tolerability and pharmacokinetics of DRV/r QD in treatment-na·ve adolescents, 12 to <18 years
|
| |
| |
Reported by Jules Levin
6th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, 17-20 July 2011
Patricia Flynn,1 Stephane Blanche,2 Carlo Giaquinto,3 Svitlana Komar,4 Antoni Noguera-Julian,5 Steven Welch,6 Erkki Lathouwers,7 Tom Van de Casteele,7 Peter Vis,7 Magda Opsomer7
1St. Jude Children's Research Hospital, Memphis, TN, USA; 2H™pital Necker-Enfants Malades, Paris, France; 3Ospedale di Padova, Padova, Italy; 4Centre, 'Clinic for Treatment of HIV-infected Children', Kiev, Ukraine; 5Hospital Sant Joan de DŽu, Universitat de Barcelona, Barcelona, Spain; 6Birmingham Heartlands Hospital, Birmingham, UK; 7Tibotec BVBA, Beerse, Belgium

FDA = Food and Drug Administration; RAM = resistance-associated mutation; AEs = adverse events
Intoduction
Sekar V, et al. 9th IWCPHIV 2008. Abstract P42
AUC = area under the concentration-time curve; C0h = predose concentration; EC50 = effective concentration
Introduction
· The protease inhibitor (PI) darunavir, in combination with low-dose ritonavir (DRV/r) and other antiretrovirals (ARVs), is approved once and twice daily for the treatment of ARV-na·ve and -experienced HIV-1-infected adults and twice daily in ARV-experienced paediatric patients aged 6 to <18 years.1,2
· Twice-daily DRV/r (weight-based dosing) has been assessed in treatment-experienced children aged 3 to <6 years and 6 to <18 years in the Phase II ARIEL (TMC114-C228; dArunavir in tReatment experIenced pEdiatric popuLation; clinicaltrials.gov identifier NCT00919854) and DELPHI (TMC114-C212; Darunavir EvaLuation in Pediatric, HIV-Infected, treatment-experienced patients; clinicaltrials.gov identifier NCT00355524) trials, respectively3,4
- in the ARIEL study, at Week 24 with DRV/r, 55.6% of patients achieved the primary efficacy endpoint of HIV-1 RNA <50 copies/mL (intent-to-treat/time-to-loss of virological response [ITT-TLOVR])3
- in DELPHI, DRV/r also demonstrated efficacy (48% of patients achieved HIV-1 RNA <50 copies/mL; TLOVR) and favourable safety and tolerability over 48 weeks.4
· Once-daily DRV/r 800/100mg has been assessed vs lopinavir/r 800/200mg (total daily dose) over 192 weeks in treatment-na·ve adults in the Phase III ARTEMIS (TMC114-C211; AntiRetroviral Therapy with TMC114 ExaMined In Na·ve Subjects; clinicaltrials.gov identifier NCT00258557) trial5
- DRV/r provided sustained and durable virological and immunological responses; at Week 192 68.8% of patients achieved HIV-1 RNA <50 copies/mL in the DRV/r arm vs 57.2% in the lopinavir/r arm (difference in response: 11.6% [95% confidence interval (CI): 4.4-18.8])5
- DRV/r was associated with fewer discontinuations due to adverse events (AEs), smaller median increases in levels of total cholesterol and triglycerides, and a lower incidence of grade 2-4 treatment-related diarrhoea than lopinavir/r.5
· DIONE (TMC114-C230; DarunavIr Once-daily in treatment-Na·ve adolEscents; clinicaltrials.gov identifier NCT00915655) is a Phase II, 48-week, open-label trial of once-daily DRV/r 800/100mg co-administered with zidovudine (AZT)/lamivudine (3TC) or abacavir (ABC)/3TC in treatment-na·ve, HIV-1-infected adolescents aged 12 to <18 years.
· Efficacy, resistance, safety, tolerability and pharmacokinetic (PK) outcomes from the primary 24-week analysis of DIONE are presented.
Methods
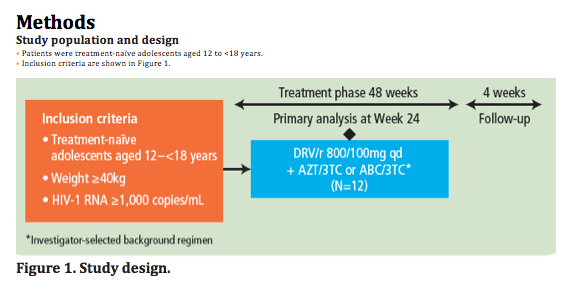
Study population and design
· Patients were treatment-na·ve adolescents aged 12 to <18 years.
· Inclusion criteria are shown in Figure 1.
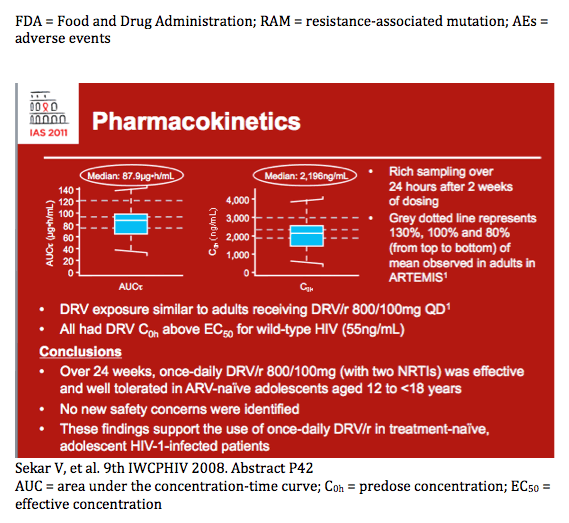
Figure 1. Study design.
· The primary objectives were to evaluate the efficacy, safety, tolerability and pharmacokinetics of once-daily DRV/r 800/100mg in combination with an investigator-selected background regimen (consisting of either AZT/3TC or ABC/3TC) over 24 weeks.
· Safety and efficacy were assessed at screening, baseline, Weeks 2, 4, 8, 16 and 24 and every 8 weeks up to Week 48.
· Patients benefiting from treatment could continue therapy in a rollover programme.
· The study protocol was reviewed and approved by the appropriate institutional review board or ethics committee(s) and health authorities, and the study was conducted in accordance with the Declaration of Helsinki. Parents or legal representatives and patients (where appropriate, depending on age and local regulation) had to be willing and able to give consent.
Efficacy, resistance and safety analyses
· Efficacy was assessed using the ITT-TLOVR algorithm
- the primary efficacy endpoint was HIV-1 RNA <50 copies/mL at Week 24.
· For resistance analyses, phenotypic and genotypic determinations were performed on plasma samples by means of Antivirogram¨ and population sequencing, respectively, at baseline, Week 24, Week 48 or at the time of early withdrawal (before Week 48) and at additional visits for virological failures (VFs)
- VF was defined as patients who achieved and then lost (rebounders) or never achieved (never suppressed) HIV-1 RNA <50 copies/mL (TLOVR non-VF censored algorithm).
· Safety was assessed using the ITT population
- the incidence and severity of treatment-emergent AEs and laboratory abnormalities was assessed. Causality was assessed by the investigator.
Pharmacokinetic analyses
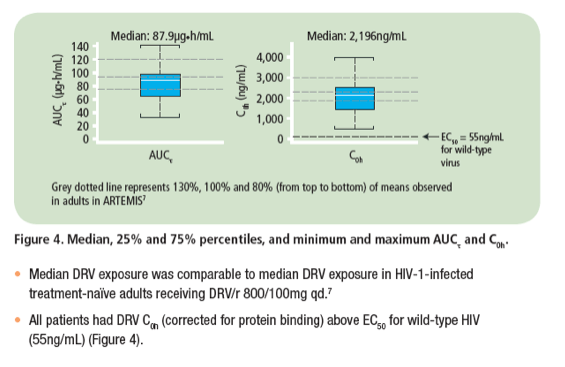
· For PK analyses, rich sampling was performed over 24 hours after 2 weeks and sparse sampling after 4 and 24 weeks of dosing
- plasma concentrations of DRV were determined using a validated liquid chromatography-mass spectrometry/mass spectrometry method with the lower limit of quantification of 5.00ng/mL
- PK over 24 weeks was assessed using a population PK model in NONMEM.
Results
Patient disposition and baseline characteristics
· Of the 12 patients who were screened, all 12 were enrolled and treated with DRV/r 800/100mg qd plus AZT/3TC (n=6) or ABC/3TC (n=6).
· No patients discontinued therapy during the 24 weeks.
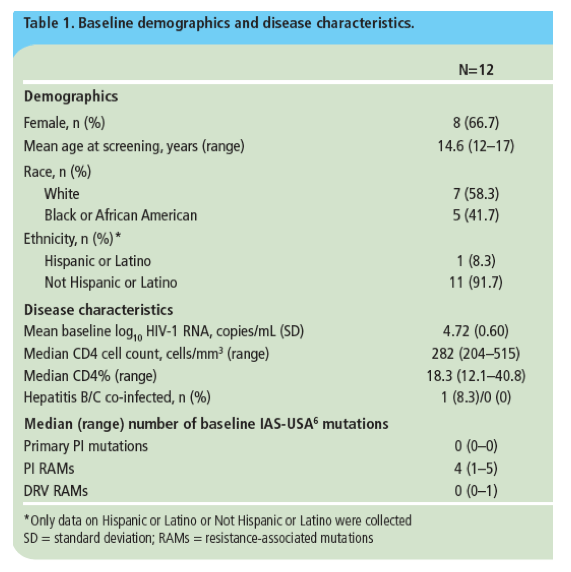
· Baseline demographics and disease characteristics are shown in Table 1.
· Modes of HIV transmission were: mother to child transmission (n=5 [41.7%]), heterosexual contact (n=3 [25.0%]), blood transfusion (n=1 [8.3%]) and other (n=3 [25.0%]).
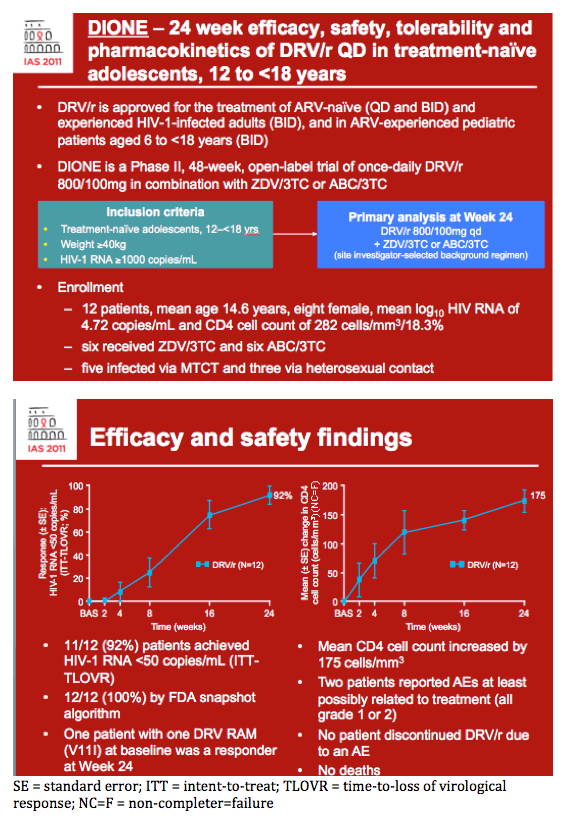
Efficacy
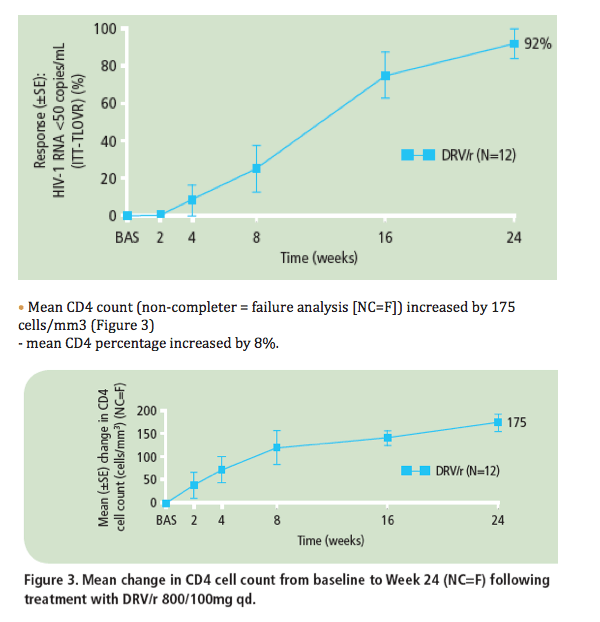
· At Week 24, 11/12 (92%) patients achieved HIV-1 RNA <50 copies/mL (ITT-TLOVR) (Figure 2)
- the remaining patient had an unconfirmed HIV-1 RNA value <50 copies/mL at Week 24
- all 12 (100%) patients achieved HIV-1 RNA <50 copies/mL according to the FDA snapshot algorithm.
· All 12 patients achieved HIV-1 RNA <400 copies/mL (ITT-TLOVR).
Figure 2. Proportion of patients achieving confirmed HIV-1 RNA <50 copies/mL (ITT-TLOVR) over 24 weeks following treatment with DRV/r 800/100mg qd.
Virology
· One patient with one DRV RAM (V11I) at baseline was a responder (HIV-1 RNA <50 copies/mL; ITT-TLOVR) at Week 24.
· One patient was considered a VF at Week 24 (TLOVR non-VF censored algorithm)
- this patient's viral load was never suppressed in the analysis as the patient had achieved an unconfirmed HIV-1 RNA <50 copies/mL at Week 24, but not two consecutive undetectable values within the 24-week observation period, as required by TLOVR algorithm to determine success.
· No post-baseline genotypes/phenotypes were available at the time of the Week 24 analysis.
Safety
· The most frequently reported AEs (reported in ≥2 patients, regardless of causality or severity) were anaemia (n=3), nausea (n=3), vomiting (n=3), cough (n=2), diarrhoea (n=2), furuncle (n=2), pyrexia (n=2) and sinusitis (n=2).
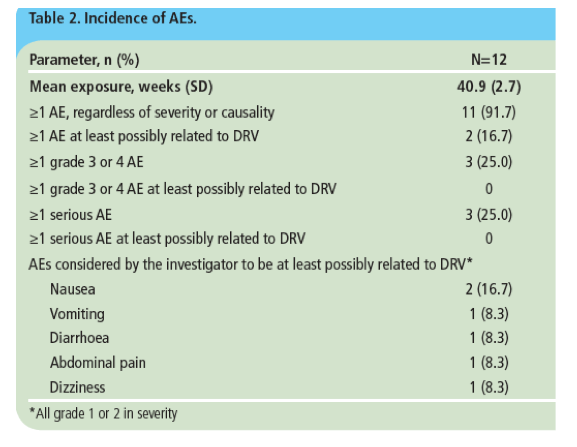
· Two patients reported ≥1 AE considered by the investigator to be at least possibly related to treatment (Table 2)
- all treatment-related AEs were grade 1 or 2 in severity.
· No patients discontinued treatment due to an AE.
· No fatal AEs occurred during the trial.
· Three patients presented with a grade 3 or 4 AE during the treatment period. These were anaemia (in two patients), leucopenia, neutropenia, diarrhoea, nausea, vomiting and hypotension (each in one patient)
- both patients who reported grade 3 or 4 anaemia had grade 1 anaemia in their medical history and were also receiving AZT
- all haematological events were considered by the investigator to be related to AZT, and improved after switch in background regimen to tenofovir/emtricitabine (n=1) and ABC/3TC (n=1).
· Serious AEs were reported in three patients and were anaemia (n=2), neutropenia (n=1), and cervical dysplasia (n=1).
Laboratory safety
· Grade 3 or 4 laboratory abnormalities were observed in two patients and reported as AEs: grade 3 white blood cell count decreased (n=1), grade 4 segmented neutrophils decreased (n=1) and grade 4 haemoglobin decreased (n=2).
· Grade 2-4 treatment-emergent lipid-related laboratory abnormalities were observed for total cholesterol, low-density lipoprotein and hyperglycaemia; no grade 2-4 increases in triglycerides were observed (Table 3).


|
| |
|
|
|
|
|