 |
|
|
| |
HIV Latency and Eradication at CROI 2012
|
| |
| |
David Margolis, MD University of North Carolina
At last year's CROI, the word "Cure" was spoken aloud with at a number of sessions and presentations. This year the topic hasd grown to seem worthy of its own category. With the spreading realization that we cannot treat all the patients in need with lifelong ART, and growing funding for research in the area, it seemed (to this admittedly biased author) an area of intense interest at the meeting. And most exciting, given the presentations made, it seemed that one could actually see the first progress being made towards the distant goal of cure, and that it was conceivable that the challenges could someday be overcome.
However, to discuss cure or eradication, we should first define terms. When people are HIV-infected, they have HIV particles in their bodies, produced by infected cells, containing HIV RNA, and these viruses are able to infect new cells, new people, and cause disease either directly by killing cells or by causing inflammation. When HIV infects a cell, it is copied into DNA and integrates into the host cell, essentially becoming a new gene in the genome of the infected person. Many of these viral genomes are defective and cannot make a virus, but even the defective ones might make HIV proteins, cause inflammation, and induce some sort of disease.
"Cure" I think implies the hope for a state in which one must take no drugs, will have a healthy immune system, is not infectious to anyone (including their children), and will not get any illness related to having had HIV in their body --- either an immune deficiency or an inflammatory problem. To achieve this it may be necessary to remove, destroy, or inactivate of all the virus and functional viral genomes in a person. Perhaps some viral DNA genomes might be left behind, as long as they were not doing anything that causes any damage or inflammation. This is a pretty tall order, but I think it is what we are talking about when we talk about "cure" or "eradication."
I have to say that I am skeptical of the utility of the alternate goal, called "functional cure." In this state infectious HIV is still present in the body, but is tightly controlled by the immune system, or by immune cells resistant to infection, without the use of medications. Such a person would have a normal healthy immune system, and no "inflammation problems." There would be no chance, or very little chance, of transmission of infection. This seems to me to be even a taller order, as we know that even natural elite controllers of HIV infection may transmit HIV, and such patients tend to have elevated levels of inflammatory markers, and do not have totally normal immune systems. To me it seems that total impact on the health of the individual of such a low-level infection, controlled infection, are not likely to be better than the impact of lifelong drugs on the health of the individual, and might be worse. But this is an opinion, and we need more research to move towards the goal of life without chronic ART, one way or the other.
At CROI 2012, presentations addressing HIV persistence were found at the Wednesday afternoon Symposium (Session 33), a Thursday morning oral abstract presentations (session 42), and several poster presentations, some highlighted at the Thursday afternoon themed discussion (Session 48). These can all be seen at www.retroconference.org
Sharon Lewin (abstr. 106 sess 33, Burnet Inst, The Alfred Hosp and Monash Univ, Melbourne, Australia) opened the Symposium with an overview talk. She reviewed the major barriers to cure that, subject to some debate, she delineated as latently infected cells, low-level persistent virus replication, and anatomical reservoirs. She reviewed data that is interpreted to mean that persistent immune activation and cell-cell transfer of virus may facilitate virus persistence in patients on ART, especially in selected tissues such as the gastrointestinal tract. Various studies have detected HIV RNA in plasma or tissue of patients on ART, and this is often called residual replication. I would prefer to call such events measured as persistent viral expression. It is incontrovertible that there must be some cells, somewhere in patients clinically suppressed on ART that are producing viral particles that contain HIV RNA. However, as numerous studies have not demonstrated evolution of viral sequences on ART, the development of drug resistance on stable ART, or changes in circulating HIV RNA or cell-associated HIV DNA with ART intensification, there is still no definitive evidence that there are ongoing full cycles of viral replication on stable ART.
webcast link Session 33 (of note is to listen to Courtney Fletcher's talk where he describes research finding reservoirs where ARTs do not reach impacting on potentially curing HIv if HAART is needed):
http://app2.capitalreach.com/esp1204/servlet/tc?c=10164&cn=retro&s=20481&&dp=player.jsp&e=16634&mediaType=podiumVideo
Lewin cited a recent paper from the laboratory of Nobel-prize winner David Baltimore (Sigal et al., Nature 2011) that showed evidence of the potential of cell-cell transmission of HIV despite the presence of antivirals. However, the paper reported to "explore a novel mechanism for ongoing HIV replication in the face of antiretroviral drugs....propose a model whereby multiple infections per cell lead to reduced sensitivity to drugs without requiring drug-resistant mutations, and experimentally validate the model using multiple infections per cell by cell-free HIV in the presence of the drug tenofovir....Infections originating from cell-free virus decrease strongly in the presence of antiretrovirals tenofovir and efavirenz whereas infections involving cell-to-cell spread are markedly less sensitive to the drugs....If cell-to-cell spread has the same properties in vivo, it may....potentially contribute to viral persistence." What is shown in the paper, lab models only but not in the presence of true combination ART, is far from proof of this hypothesis, but does raise the possibility.
Lewin next reviewed strategies being proposed or explored to achieve eradication or functional cure. The first of these are agents designed to perturb latent infection and induce the expression of quiescent provirus within resting CD4+ T cells. Such agents included histone deacetylase inhibitors such as SAHA or vorinostat, the cytokines IL7 or IL15, the drug Disulfiram, and methyltransferase inhibitor 5-azacytidine, NF-kappaB activators such as prostratin, and other molecules alone or in combination. SAHA and 5-aza appear to act by modifying histone proteins that are a scaffold around which the HIV genome is wrapped in a way that allow expression of the viral genome, although other effects on cellular proteins of these inhibitors of human regulatory enzymes may also play a role in HIV induction. IL7, IL15, prostratin, and Disulfiram may engage signaling pathways in the host cell that also result in expression of latent HIV.
Dr. Lewin outlined the design of a vorinostat (VOR) study that she is performing in which the endpoint is the measure of cell-associated HIV RNA in total T cells. She reported that 10 patients have received VOR for 14 days, in thus far in 9 patients she did not observe a change in CD4 counts, plasma HIV RNA at a <20 c/ml limit, or changes in T cell activation in some rectal biopsies performed. A few grade 1-2, reversible adverse events were reported.
She also outlined several other studies that I will discuss in detail later:
1. the study performed in our group at UNC, focused differently on measuring precisely the effect of a single dose of VOR on expression of HIV within a latent reservoir -- resting CD4+ T cells in the blood - in real time (abstract 157LB and weblink).
2. A study of the drug Disulfiram
3. Studies of PD-1 receptor blockade via anti-PD1 antibody
4. A gene therapy study attempting to alter the CCR5 receptor and create HIV-resistant CD4 cells
Dr. Lewin closed with a summary of the progress towards eradication or functional cure thus far. So far, in contrast to early attempts to eradicate infection via global activation of CD4 T cells, thus far initial pilot studies have been safe and well-tolerated. Thus far no intervention tested has resulted in a convincing increase of plasma viremia, even at low levels. No evidence of immune activation or damage has bee noted thus far. However, the field is still challenged by the difficulties inherent in measuring events relevant to rare latent, persistent infection, the only convincing metric --- therapy interruption --- is uniquely challenging, and the most convincing assay --- careful quantitation of latent, resting CD4 T Cell infection --- is costly and cumbersome. Clinical trials that test strategies for HIV eradication pose multiple unique challenges, including the need for more accurate and standardized assays to quantify persistent virus in patients on cART, identification of the most appropriate clinical end points for eradication studies, including the role of treatment interruption, and very careful consideration of the risk benefit for any intervention.
Next Jeff Lifson (NCI-Frederick, MD, abstr. 107) provided an overview and opinions on the role of non-human primate models in the eradication research. Given the potential risk of such research in the clinic, particularly in patients for whom there is little to no likelihood of immediate benefit, non-human primate models can play an important role eradication research. However, Lifson outlined practical limitations that still leave non-human primate as an imperfect tool. Several different macaque species (usually rhesus or pigtail macaques) are in use in different laboratories. Even the type of virus differs widely. Studies utilize simian immunodeficiency virus [SIV] isolates of differing virulence and cell tropism, while others use a chimeric human-simian virus (reverse transcriptase-simian-human immunodeficiency virus [RT-SHIV]) that can be treated with human RT inhibitor drugs. The field is not yet developed enough to reliably infect and durably suppress viremia with the same profound success as can be done in human infection. Finally, SIV in non-human primates may not completely and fully represent HIV pathogenesis in humans.
Lifson showed some of his own data from an ongoing study attempting, as in the two ongoing human studies, to measure the effect of the HDAC inhibitor VOR on latent infection in ART-treated monkeys. Suppression of viremia to <30 copies/ml was reported, but remarkably this required the use of PMPA (the prodrug of tenofovir), FTC, two integrase inhibitors and ritonavir-boosted darunavir. Additionally, Lifson showed that he could measure the biomarker of affect of VOR --- histone acetylation --- in PBMCs of monkey that were dosed with VOR. In ex vivo studies (monkey cells in the lab, outside the monkey) he showed that VOR could disrupt latency of SIV infection. Proof of this in vivo in monkeys is awaited.
Lifson made the point that virologic monitoring in non-human primate models benefits from the ability to obtain multiple, carefully timed specimens, including extensive tissue specimens, although blood volumes can be limiting. All such studies can also be done in parallel with careful measures of plasma and tissue drug levels. A new and sensitive PCR technique called "digital PCR" may allow for the detection of rare cells with inducible viral RNA. Therefore, while the technology is not yet perfected, non-human primate studies seem certain to play a key role in the addressing important questions about persistence of HIV infection in the face of ART, and single and multi-modality approaches to cure or functional cure.
Courtney Fletcher (abstr. 108, U. Nebraska Medical Center) then reprised his presentation that was first made at the St. Martin Persistence workshop in December, and then reported as News & Views piece in Science magazine. This work is still in preliminary form, and has not yet been peer-reviewed or published, but already appears to be exerting a powerful influence on thinking in the field. The hypothesis put forward is that cryptic replication, that is full rounds of the HIV lifecycle -- entry, reverse transcription, integration, viral gene expression, virion production -- occurs continuously or intermittently in tissue such as the lymph nodes of GALT is driven by low ART concentrations in the lymph nodes and GALT. I personally do not understand how this would occur, as it seems that eventually a drug-resistant virus would appear at the site of replication, but would leak out to some other site. In case, eradication strategies currently depend on the concept that ART can block the vast majority of new infection events, and so it is certainly important to understand the pharmacology of different tissues. This work is an important step in that direction.
In this study, patients who initiated therapy at Tim Schaeker's clinical site at U Minn underwent blood and tissue sampling at the initiation of ART, and months 1, 3, and 6. It was said that all the patients seemed to be compliant, and had therapy success at 6 months. 9 patients of a planned total of 12 have been studied (although perhaps the planned study size has now been expanded even further). ART drugs that were assayed included the protease inhibitor atazanavir (ATZ), and RT inhibitor efavirenz (EFV) and the phosphorylated, activated forms of the RT inhibitors tenofovir diphosphate (TVF-DP) and emtricitabine triphosphate (FTC-TP). Detectable levels of all of these drugs were found in PBMC. But of note, drug concentrations were highly variable in the lymph nodes, often somewhat low (compared to target plasma concentrations) in ileal GALT tissue biopsies, and usually acceptable in rectal biopsy tissues. However, I find the data very difficult to interpret, as there are very small numbers of patients on 3 different combination regimens (Atripla or Truvada/boosted ATZ or Truvada/boosted darunavir[DRV]). It was also difficult to tell if the data reported represented different numbers of tissue samples for patients (ie one biopsy piece or the the average of multiple pieces). It was said that 9 subjects had completed all procedures, which would have included biopsies at months 1, 3, and 6, but at least some of the PBMC samples seemed to have come from perhaps more than the 9 patients. Finally it was difficult to understand what the positive controls would be: that is how much drug is expected in one piece of tissue vs. another (one tissue might have more or less connective tissue vs fat vs lymphoid tissue). With tissue being taken from different sites during a clinical procedure, processed in the GI suite and then shipped from one state to another, some sort of internal standard for the intactness of the specimen would be desirable.
In general ATV and DRV levels were undetectable in all of 9 node pieces, but detectable in most ileal and rectal biopsies. TFV-DP levels were as expected in PBMCs, but somewhat low in nodes and high in ileum/rectum. FTC-TP levels were as expected in PBMCs, but somewhat low in nodes and even lower in ileum/rectum. ATZ levels were higher than expected in PBMCs, undetectable in nodes, low in ileum and high in rectal biopsies. Efavirenz levels seemed OK in PBMCs and most biopsies but low in nodes.
With this surprising but concerning data as a backdrop, Dr. Fletcher wrapped up his talk with examples from medical history on the importance of pharmacology in the use of drugs, and improvement of various clinical outcomes when drug therapy is based on good pharmacological data. There is no doubt we need more data in this area.
Daria Hazuda (abstr. 109 session 33; Merck Res Labs, West Point, PA) then gave an overview talk of the issues and challenges that face those seeking to develop drugs or interventions that can eradicate HIV-1 infection or achieve a functional cure. The considerable challenge will likely require strategies to purge the reservoir of latently infected cells, as well as approaches that can enhance the clearance of either latently or persistently infected cells. The former, and to some extent the latter are endeavors that have no precedent or in pharma-speak no "pathway to licensure." It is unlikely that any one company really wants to "own" the cure for AIDS, and almost certainly that it will be complicated enough that parts of it will come from many sources, but on the other hand it takes millions of dollars of resources to develop anything this revolutionary.
On the positive side, increasing understanding of the molecular mechanisms and cellular reservoirs that allow latent proviral genomes to persist and infection to be maintained despite ART have suggested potential therapeutic approaches. For example it is fairly straightforward to perform unbiased, high-throughput screens of chemical libraries to seek small molecules that induce the expression of integrated HIV genomes. Over the years a hierarchy of different systems for this task has developed: cell lines encoding reporter genes driven by the HIV LTR promoter, T cell lines that better mimic true resting CD4+ T cells and encode either a LTR-driven reporter or a cloned proviral genome, primary cells infected in the laboratory and then returned to a quiescent state via cytokine manipulation, and finally difficult and costly assays of cells directly from patients.
Dr. Hazuda outlined the efforts of one such screen, using technology of small interfering RNA (siRNA). siRNAs are small RNAs that interfere with the translation of the RNA to which they are matched, and therefore "knockdown" the expression and effect of a single human gene. By creating a library of siRNAs that target every human gene transcript (or at least a lot of them), and shotgunning them into pools of target cells that are some sort of model of latent HIV infection, it is possible to find human genes that if "knocked down" result in the disruption of HIV latency. Using a Hela cell line that encodes an HIV reporter gene - something pretty far away from a latently infected patient's cell but a cell line amenable to this screening approach - Dr Hazuda's laboratory found over 400 human genes whose activity was somehow related to the maintenance of HIV latency, and whose disruption resulted in expression of latent HIV in this model system. Interestingly, this screening approach identified HDACs as an effective target, as well as several other genes. HDAC inhibitors, as has been discussed, are now in human testing for this use. However, thus far these other genes have not yielded any new drug targets for further development. New screening efforts are underway in a system thast is hoped to behave more like resting CD4 T cells and yield new drug targets.
Dr. Hazuda then discussed the second challenge that is expected to require attention, that of killing or clearing infected cells, once latency is disrupted. One approach now in animal testing is the use of anti-PD1 antibodies. PD-1 receptor that negatively regulates the immune response. Blockade of PD-1 by anti-PD1 in HIV-infected patients is hoped to augment the immune response and improve clearance of HIV-infected cells. Interferon, a cytokine with direct general antiviral effects, is also being tested. Several therapeutic vaccines, designed to allow control of viremia after ART interruption have entered clinical testing. The use of such immune augmenting strategies might be used together with agents that disrupt latency, the so-called "shock-and-kill" approach to eradication or functional cure of HIV infection.
On Thursday morning (Session 42) a variety of talks were presented in a session entitled "HIV Persistence, Latency, and Eradication." First, Maria Buzon (abstr. 151) discussed the effect of early ART on persistent infection. The group studied CD4+ cells were isolated after a median of 10 years of undetectable viremia from 9 patients treated during early/acute infection (<90 days), 27 treated in chronic infection, and 37 untreated "elite controller" patients. Total cellular HIV DNA, LTR circular DNA, HIV DNA integrants, and viral outgrowth from unsorted PBMCs was reported. As others have seen, she found a rapid decrease in HIV DNA, and the speed of this decline was not increased by earlier therapy.
In comparison to chronic treated patients, levels of integrated and total HIV-1 DNA were significantly lower in EC (p = 0.003 and p <0.0001, respectively) and patients treated during acute infection (p = 0.06 and p = 0.001, respectively). 2-LTR circles were also more frequently detected in chronic treated (5/10) than EC (3/37) (p = 0.002) and early treated subjects (1/8)(p = 0.09). The calculated half-life of integrated HIV-1 DNA in early treated patients was 61 months, consistent with the extremely long persistence of latently infected cells. So although some measures of HIV infection decay more rapidly in patients treated earlier, measures of persistent infection (DNA integrants, viral recovery) do not decay more rapidly.
It is difficult and perhaps inappropriate for me to report on a presentation for which I was a co-senior author, so I will keep this brief. Alan Perelson (abstr. 152) reported the findings of our collaborative study of measures of infection and latency in a group of patients treated in truly acute infection - within 45 days of HIV RNA+ Western blot negative diagnosis. Once HIV RNA was <50 copies/mL for >6 months, leukapheresis was performed and resting cell infection (RCI) quantitated by a co-culture assay. Low-level viremia was measured by single-copy assay (SCA). A mathematical model that predicted the frequency of latent infection based on an estimate of "area-under-the-curve of viremia from infection to ART-induced suppression, adjusted for changes in CD4 cell count, accurately predicted the frequency of latent infection (r = 0.65, p < 0.0003). While RCI declined after the first months following ART suppression of viremia in most patients, there was a striking absence of decline when initial RCI frequency was less than 0.3 per million. Low-level SCA viremia was observed in 0 of 4 patients with RCI <0.3/million, and 4 of 6 patients with RCI >0.3/million (Fisher's 2-tailed p = 0.076). This suggested that the degree of resting cell infection is directly related to the availability of CD4+ T cells susceptible to HIV, whether viremia is controlled by the immune response and/or ART, and that 2 pools of infected resting CD4+ T cells exist�less stable cells, observable largely in patients with high levels of initial viremia, and extremely stable cells that are established despite very early ART. Preliminarily, the detection of low-level viremia by SCA despite ART appears to be correlated with RCI.
Next Laing Shan (abstr. 153) reported for the Siliciano laboratory, finding that most ART-treated patients lack the effective HIV-specific CTL response needed to eliminate HIV-expressing cells. The full manuscript happened to be published on-line immediately after the meeting in the journal Immunity, a case of perfect timing. CTL response may be required for clearance of infection once reagents that disrupt latency are put in to place. Shan treated PBMCs from patient with ART and 500 nM VOR in ex vivo culture for five days, and then isolated resting CD4+ T cells that remained in the culture. HIV recovery per million cells was then quantitated by a viral outgrowth assay. There was no reduction in recovery of virus per million cells after this procedure. This was interpreted as showing that VOR-induced virus expression did not kill infected cells in culture.
Then Shan presented a different set of experiments to demonstrate killing of latently infected cells. in vitro from primary CD4+ T cells from patients on HAART. After reversal of latency, infected resting CD4+ T cells survived despite viral cytopathic effects, even in the presence of autologous CD8+ T cells from most patients on HAART. Killing of infected cells was observed only at high E:T ratios after a longer period of co-culture. Antigen-specific stimulation of CD8+ T cells before co-culture partially restored the cytolytic T lymphocyte (CTL) functions and led to efficient killing of latently infected cells. Shan concluded that resting CD4+ T cells latently infected with HIV-1 will not be killed by either viral CPE, or host CTL responses after virus reactivation. There were several technical issues with these experiments that in my opinion makes this sort of absolute statement premature. However, this is a distinction without a difference, as Shan also concluded that stimulating HIV-1-specific CTL prior to reactivating latent HIV-1 may be essential for successful eradication efforts and should be considered in future clinical trials. There is no arguing with this conclusion, as better immunity against HIV cannot possibly be a bad thing, and I cannot think of any viral infection in history that has been cleared in the absence of an immune response.
We all know that the Berlin patient was cured of HIV after multiple interventions, including a bone marrow transplant. Cillo and colleagues (abstr. 154) attempted to assess the contribution of intensive myeloablative chemotherapy and autologous hematopoietic stem cell transplantation (ASCT) to this result. In 10 HIV+ patients from 3 clinical sites who underwent ASCT. Persistent viremia was detected in 9 of 10 patients post-SCT, with a median HIV-1 RNA of 1.5 copies/mL (range: <0.2 to 26). These findings suggest that allogeneic stem cell transplantation with CCR5 defective donor cells, as was done with the Berlin Patient, was critical in achieving the reported cure of HIV.
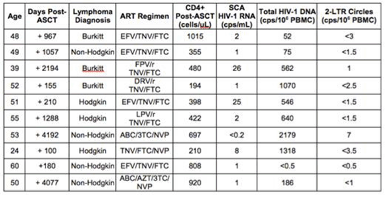
Pablo Tebas (abstr. 155) reported the finding of a gene therapy study in which CD4 cells from ART-treated HIV+ patients were treated with a designer enzyme called a zinc-finger nuclease, designed to induce a mutation and create an acquired deficiency of the CCR5 HIV co-receptor on the modified CD4 cells. Like people who naturally possess the CCR5 Δ32 mutation, an acquired deficiency of CCR5 receptor will make CD4 T cells resistant to R5 virus infection. I did not really notice any new data that was added to this presentation since it was first reported at ICAAC in the fall of 2011, but maybe I missed something. Tebas reported that this was primarily a safety study, and that the procedures and reinfusion of cell was safe. After CD4 infusion patients with low CD4 cells realized an increase of about 500 cells, and patients with higher baselines increase over 1500 cells. In most patients CD4/CD8 ratios normalized, and detection of a pentamer sequence which denoted the persistence of gene-modified cells (at least at a level that was detectable by PCR) also persisted for up to a year. In most patients who underwent a planned ART interruption after cell transfer, HIV RNA rebound was somewhat lower than original setpoint at about 140 days. In one patient who already had one copy of the CCR5 Δ32 mutation, rebound viremia was still undetectable after 2 weeks of interruption. These are initial findings of safety and feasibility, but how this approach will be translated to clinical utility remains to be seen. The researchers have proposed to treat patients with mild chemotherapy designed to kill off some of the bone marrow, and "make room" so that a larger percentage of modified CD4 cells can be transferred.
Sifei Xing (abstr. 156) then presented the results of a chemical library screen performed in the Siliciano laboratory for compounds that reactivate latent HIV-1 in a Bcl-2 transformed primary CD4+ T cell model. She found that a group of quinolin-8-ol derivatives that can induce latent HIV-1 in this model without causing global T cell activation, and that none of the analogs induced cytokine secretion in primary resting CD4+ T cells over 48 hours. Further testing will be required to demonstrate that these compounds will serve as a platform for the development of safe and effective reagents to perturb latent infection in the clinic.
This screening system previously indentified Disulfiram as a drug that could induce the expression of latent provirus. However, Disulfiram did not induced the expression of HIV from patient's cells in an ex vivo outgrowth assay (Xing, J Virology 2011). Therefore Spivak undertook a pilot human study to test the activity of Disulfiram in vivo (abstr. 369). Disulfiram was given at 500 mg for 14 days. There was no change measured in the frequency of resting CD4 cell infection by an outgrowth assay. This study is being done at UCSF and Hopkins, and only at Hopkins was single copy viremia measured within 2 hours of Disulfiram dose. In one patient there was an increase in viremia from 15 to 65 copies at 2 hours, and modest increases of about 10 copies/ml in two of 7 other patients.
Several other screening programs were presented during the poster session. Ramsussen (Aarhus Univ Hosp, Denmark; poster 370) found that the HDAC inhibitor panobinostat (LBH589) stimulates HIV-1 expression more potently in the latently infected T-cell line ACH2 and the monocytic cell line U1 than other HDACis such as ITF2357 (givinostat), PXD101 (belinostat), SAHA (vorinostat), and valproic acid (VPA). These findings require further investigation for several reasons: cell lines are crude models of latency, and while many findings in these models are relevant, probably more are not. Secondly, "potency" in the disruption of latency may be irrelevant, or even harmful. All that matters is that a drug can achieve a level in vivo that is safe and effective. A less potent drug will work if more milligrams can be given. It is also not know how "potently" one should induce latent expression, the worst example being the OKT3 study of the 1990's in which patient were harmed by overly potent T cell activation.
Bosque of Utah (abstr. 366) presented more advanced system in primary in vitro differentiated central memory cells. How to use advanced systems such as this to more effectively identify anti-latency reagents is an important research priority.
This writer then presented the findings of our group in the first reported clinical test of the ability of a single oral dose of the HDAC inhibitor vorinostat (or SAHA) to disrupt HIV-1 latency in patients on ART (abstr. 157LB). I refer you to the report of David Schepp for his impression of our study.
CROI 2012: HIV Persistence, Latency and Eradication - written by David H. Shepp, MD Associate Professor of Medicine Hofstra North Shore-LIJ School of Medicine
North Shore University Hospital - Manhasset, NY (03/19/11)
Finally, Vijayakumar Velu of Emory presented the results of a study of anti-PD-1 after ART Interruption in SIV+ Macaques (abstr. 158LB). Monkeys were treated with ART at 16 weeks post infection for 21 weeks, after which the therapy was interrupted. Four weeks post ART interruption, 6 out of the 10 macaques were treated with 6 doses of fully humanized anti-PD-1 antibody (3mg/kg) on days 0, 3, 7, 14, 21, and 28, and the remaining 4 animals were left untreated (controls). All macaques were monitored for the immune restoration and viral control. Following ART interruption, the plasma viremia increased rapidly in all animals. PD-1 blockade 4 weeks after ART interruption resulted in decline of plasma viral load in 3 out of the 6 animals. PD-1 blockade did not enhance the frequency of SIV-specific CD8 T cells but rather enhanced the functional phenotype as measured by the enhanced expression of the T cell co-stimulatory marker CD28. If the use of anti-PD1 can be shown to be safe in humans, it may provide a tool to enhance immune response and control of eliminate HIV infection.
Overall, there was a huge increase in activity and interest in latency and eradication at 2012 CROI. One can hope for more real progress by 2013.
|
| |
|
|
|
|
|