 |
|
|
| |
Clinical Pharmacology at the 13th Workshop on Clinical Pharmacology of HIV Therapy
|
| |
| |
(Clinical Pharmacology of Hepatitis C Therapy and Drug-Drug Interactions; Drug-Drug Interactions - darunavir/cobi, GS-7340/cobi, elvitegravir/cobi-reyataz, dolutegravir, TB meds, efavirenz)
".......the message is: we are at or beyond our ability to confidently predict interactions (two-, three-way or more) among ARVs and HCV PIs, which are all substrates, inhibitors and inducers of drug metabolizing enzymes and membrane transporters. If there are not PK data that provides support for using these combinations, I would not recommend it." see discussion below.....
13th International Workshop on Clinical Pharmacology of HIV Therapy
Barcelona, Spain
April 16-18, 2012
The 13th International Workshop on Clinical Pharmacology of HIV Therapy was held in Barcelona, Spain from April 16-18, 2012. This meeting continues to be a premier meeting focused on clinical pharmacologic issues in the treatment of HIV infection. Furthermore, it remains a venue for scientific interactions among scientists from academia, pharmaceutical industry and regulatory authorities, which is an almost unique attribute. In this report I will highlight abstracts focused on pharmacologic issues I think are of broad interest or might benefit from some expert clarification. I will discuss abstracts in four broad categories: compartmental penetration of ARVs; dose optimization, clinical pharmacology of new drugs and new combinations; drug interactions; and clinical pharmacology of HCV therapy and drug interactions.
Abbreviations
%CV, percent coefficient of variation
ABC, abacavir
ACTG, adult AIDS clinical trials group
APV, amprenavir
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
ATV, atazanavir
BOC, boceprevir
Cmin, minimum drug concentration
CNS, central nervous system
COBI, cobicistat
CSF, cerebrospinal fluid
Ctrough, concentration immediately before the next dose
CYP, cytochrome P450 drug metabolizing enzymes
DTG, dolutegravir
DRV, darunavir
ddI, didanosine
EFV, efavirenz
EVG, elvitegravir
FTC, emtricitabine
ETR, etravirine
fAPV, fosamprenavir
IC50, concentration of drug required to inhibit viral replication in vitro by 50%
IDV, indinavir
IM, intramuscular
IQ, inhibitory quotient
3TC, lamivudine
LPV, lopinavir
MVC, maraviroc
NVP, nevirapine
NRTI, nucleoside reverse transcriptase inhibitor
NNRTI, non-nucleoside reverse transcriptase inhibitor
PACTG, pediatric AIDS clinical trials group
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamic
PG, pharmacogenetics/pharmacogenomics
P-gp, p-glycoprotein
PK, pharmacokinetic
PI, inhibitor of HIV protease
PrEP, pre-exposure prophylaxis
r or RTV, ritonavir
RAL, raltegravir
RBT, rifabutin
RBV, ribavirin
RPT, rifapentine
RIF, rifampin
RPV, rilpivirine
SQV, saquinavir
SC, subcutaneous
TDF, tenofovir disoproxil fumarate
TFV, tenofovir
TVR, telaprevir
TDM, therapeutic drug monitoring
TPV, tipranavir
TB, tuberculosis
ZDV, zidovudine
The 13th Workshop opened with a superb lecture on caring for HIV-infected persons in Spain during the current economic crisis given by Bonaventura Clotet, M.D., Ph.D.. Spain is in an economic crisis; the unemployment rate is 24%. As part of government cost cutting measures, HIV units are forced to immediately reduce their ARV budgets by 10% (in Spain, ARVs are supplied through hospital pharmacies). At Dr. Clotet's unit, care is provided for 3000 patients of whom 2400 are receiving ARV therapy. Expenditures for ARVs represent 47.8% of the hospital pharmacy budget, at approximately $24 million dollars/year (in 2011). The 10% reduction in this one unit translates to a $2.4 million reduction. Three strategies were identified: (1) enrollment of more patients into clinical trials, (2) price reductions of ARVs by the pharmaceutical manufacturer, and (3) cost cutting measures, primarily changes in treatment regimens, in patients receiving ARV therapy. Dr. Clotet discussed cost cutting measures that were identified and lines of evidence to support them: changes to PI/r monotherapy, switching TDF/FTC to ABC/3TC and EFV to NVP, and withdrawing high cost drugs such as ETR and RAL. While rebates from pharmaceutical manufacturers have been the main source of cost saving to date, these cost cutting measures did result in a reduction in ARV expenditures. This was a sobering lecture to hear; it won't be the last as the economic crisis continues to play out globally. I worry about our ability to preserve the gains in life expectancy that have been achieved with ARV therapy and extend these benefits to those HIV-infected persons who need treatment, but are not yet receiving it.
Two other lectures on challenges in the management of the HIV-infected person were given by Esteban Martinez, M.D., and Roger Paredes, M.D., Ph.D.. Dr. Martinez discussed the complications and adverse reactions associated with ARV therapy, and Dr. Paredes the management of the high viral load patient and whether one dose (of ARVs) fits all patients. Both presentations were outstanding. Important take home points from Dr. Martinez on adverse reactions associated with contemporary ARV therapy were: (1) short-term adverse events (e.g. GI intolerance) are scarce and usually predictable; (2) long-term adverse events (e.g. lipodystrophy, cardiovascular, renal) are also infrequent but identifying which individuals will develop them is more difficult; (3) co-morbidities are increasingly apparent with increasing age; (4) ARVs can complicate co-morbidities; and (5) ARV therapy should be selected based on long-term efficacy and the lowest potential for adverse reactions and complications. Dr. Paredes reviewed the management of the patients with a high viral load and data on potency of ARV regimens. He concluded that for the most part, the same dose of ARVs does fit for the person with a high viral load when suppression of plasma HIV-RNA is the goal; he noted that there still remains little data in other compartments and that there are considerations other than just suppression of plasma HIV-RNA.
I. Compartment Penetration of Antiretroviral Agents
Raltegravir concentrations in gut-associated lymphoid tissue are higher than those in blood.
Patterson and colleagues presented important work (abstract O_11) on the concentrations of RAL in gut lymphoid tissue (GALT). The GALT along with secondary lymph nodes (LN) are the primary site of HIV replication and where the latent pool of virus is maintained. HIV replication in these tissues causes inflammation, immune activation and T cell depletion. Thus, knowledge of whether sufficient concentrations of ARVs reach these compartments is essential to develop effective PrEP strategies, because HIV migrates rapidly to the GALT after primary infection, and for cure strategies, because of the latent reservoir. In this study 14 male healthy volunteers received RAL, 400 mg twice daily, and underwent colonoscopy and PK studies on two occasions, after the first dose and 7 days later. After the single dose, the ratios of RAL concentrations (expressed as the AUC) of tissue to blood were: terminal ileum, 99; splenic flexure, 70; and rectal tissue, 39. After multiple dosing, these ratios were: terminal ileum, 84; splenic flexure, 679; and rectal tissue, 239. These data indicate that even after a single dose RAL concentrations in these tissues are several folds higher than in blood, and thus provide a pharmacologic rationale for studies in pre- and post-exposure prophylaxis and in primary infection. Additionally, these data add to accumulating knowledge that ARV concentrations in blood are not predictive of ARV concentrations in tissues.
The long-acting (LA) rilpivirine formulation achieves concentrations in cervicovaginal fluid and rectal tissue in males equivalent to plasma concentrations.
Else and colleagues presented an update of their work (abstract O_12) with LA rilpivirine (RPV) that was reported at CROI-2012. In this study, 10 female and 6 male volunteers received a single 600 mg IM injection of LA-RPV and concentrations in plasma were measured for 84 days. RPV concentrations in cervicovaginal fluid (CVF) and vaginal tissue (VT) were measured in female participants and concentrations in rectal fluid (RF) and rectal tissue (RT) were measured in males. The following table presents the RPV 84-day AUC in blood, and ratio of the other compartments to blood.
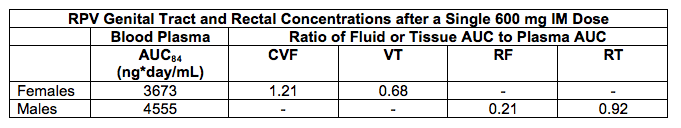
These data indicate that CVF and RT concentrations are approximately equivalent to that in plasma, whereas VT is lower, and RF considerably lower than plasma. These data provide a pharmacologic basis for additional PK and safety studies, particularly to determine concentrations after multiple dosing. But, I am uneasy that these concentrations will provide a high level of protection against HIV acquisition, based upon what we are learning about the concentrations of NRTIs required for PrEP efficacy (see Kashuba A, Lancet 2011, Dec 6 epub, and Anderson P, Abstract 31LB at CROI 2012).
Pharmacologic considerations for HIV prevention.
Peter Anderson gave an outstanding lecture on this topic. I thought the following were particularly important points:
· For PrEP, the response measure in the drug concentration-response relationship is YES or NO, which is quite different from therapeutics, where it may be the concentration associated with a 1 or 2 log drop in HIV-RNA;
· The PKPD relationship for prevention of HIV infection is likely unique to prevention and different from that for treatment;
· The PKPD relationship may be different for women vs. men-having-sex-with men (MSM);
· Drugs must reach effective concentrations in a very short time, likely within 24 hours, because of the speed with which virus can infiltrate lymph nodes; and
· Efficacy, such as that observed in the iPrEx study, may be a function of high mucosal concentrations as well as effective systemic concentrations.
Adherence.
Bernard Vrijens followed with an equally outstanding talk on understanding, describing and quantifying medication adherence. The necessity of a high level of medication adherence is nothing new to the antiretroviral therapy field. It is an even more important for the PrEP field because, as mentioned above, efficacy is either yes or no. The most recent illustration of the role of adherence was in the FEM-PrEP trial presented at CROI 2012 (abstract 32LB), which was discontinued for futility in April 2011, with 33 infections in the TDF/FTC group compared with 35 in the placebo. Adherence based on measured concentrations of TFV in plasma was < 26%. These findings lead to the conclusion that TDF/FTC likely did not work because of suboptimal adherence. A excellent review paper on medication adherence can be found here: Blaschke TF: http://www.ncbi.nlm.nih.gov/pubmed/21942628
II. Clinical Pharmacology of New Drugs and Combinations
Cobicistat Increases TFV Concentrations when given in a single tablet formulation of GS-7340, cobicistat, elvitegravir and emtricitabine.
GS-7340 is a prodrug of TFV that achieves higher intracellular concentrations from lower plasma concentrations, and therefore is more potent than TDF. For example, the IC50 for TFV is 1.2 μM, 0.015 μM for TDF, and is 0.003 μM for GS-7340. The PK of TFV following administration of the TFV prodrug GS-7340 in a single tablet formulation of GS-7340, COBI, EVG and FTC, were evaluated in a series of healthy volunteers studies (abstract O_13 and see also, http://www.natap.org/2012/pharm/Pharm_13.htm ). When GS-7340 was given in combination with COBI only, compared with GS-7340 alone, the AUC, Cmax and Cmin of TFV were all increased 3-fold, providing data that indicate an inhibitory interaction between GS-7340 and COBI. This interaction appears to be the result of COBI inhibiting the P-gp mediated intestinal efflux of GS-7340, which results in increased bioavailability of GS-7340 and therefore increased plasma concentrations of TFV. The single tablet formulation with a reduced dose of GS-7340 to 10 mg, with COBI (150 mg), EVG (150 mg) and FTC (200 mg) achieved concentrations of GS-7340 equivalent to administration of GS-7340 at a dose of 25 mg given without COBI (AUC ratio of +COBI to -COBI = 0.91) and achieved TFV plasma concentrations that were slightly higher (AUC ratio of +COBI to -COBI of 1.24). Because administration of TFV as the GS-7340 prodrug achieved higher intracellular concentrations of TFV-diphosphate (the pharmacologically-active moiety) than an equivalent dose of TDF, plasma TFV concentrations are actually lower (approximately 90%) with GS-7340 than with TDF.
I think GS-7340 is a very intriguing compound. The enhanced lymphatic delivery and intracellular concentrations of active drug (TFV-diphosphate) may represent an important next step in HIV therapeutics: improving the delivery of drug to where you want it to enhance efficacy while reducing concentrations of drug where you don't want it to improve safety. The proof of this concept of course requires comparative data on safety and efficacy. A Phase II study comparing EVG/COBI/FTC/GS-7340 with EVG/COBI/FTC/TDF (QUAD) is now underway.
DRV PK in a fixed dose combination tablet with COBI are similar to those when given with RTV.
Abstract O_20 evaluated two different fixed dose combination tablet formulations of DRV with COBI in healthy volunteers (see also, http://www.natap.org/2012/pharm/Pharm_09.htm ). In the table below, I'll only provide data on the G004 formulation as that is the one selected for further development, although the PK of these two formulations appear bioequivalent.
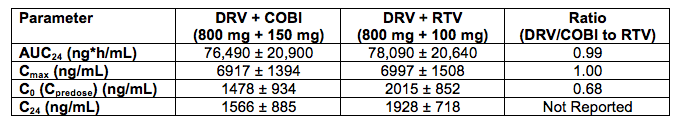
The AUC and Cmax of DRV when given with COBI or RTV are essentially the same. However, predose and 24 hour post dose concentrations, are approximately 32% lower on average with COBI than RTV. The abstract authors concluded this lower trough concentration was not clinically relevant. However, the study was done in healthy volunteers, in a fed state, in a rigorously controlled clinical environment. No data are available in HIV-infected persons, taking a different type of meal or no food at all, and with other common concomitant medications. Poster P_31 reported higher DRV trough concentrations when given once daily with RTV in the morning versus once daily with RTV in the evening (a circadian variation), indicating the time of drug administration is another variable that may affect concentrations. Jonathan Schapiro, M.D. made the point during the question and answer sessions that the burden of proof rests with the investigators to show these lower concentrations are truly not clinically relevant. I agree.
III. Drug-Drug Interactions
EFV and Rifampin: to Increase the EFV dose or not?
In December 2011, the FDA modified efavirenz (EFV) dosing in the label to recommend an increase in the dose of EFV to 800 mg once daily in individuals who are taking concomitant rifampin (RIF) and weigh more than 50 kg. This recommendation has stirred some controversy. The CDC, for example, has not yet adopted this dosing recommendation (see http://www.cdc.gov/tb/publications/guidelines/TB_HIV_Drugs/rifampin_therapy.htm ).
Abstract O_01 examined the PK of EFV in 40 children with TB after at least 2 weeks of concomitant EFV and RIF therapy. The PK were repeated at least one month after RIF was stopped. The average mid interval EFV concentration with concomitant RIF was 1.64 mg/L compared with 1.96 without RIF. These data indicate that overall, the reduction in EFV from treatment with RIF was not statistically significant in this small sample size (p=0.64), and as such they would not support an EFV dose increase. But, there are some caveats and an emerging consideration. First, the caveats. Children <20kg had an increased rate of EFV concentrations <1 mg/L. (Note, the WHO 2010 EFV dosing guidelines recommend a higher EFV dose for children between 14 to <20 kg than did the 2006 guidelines, which were used in this study.) There was an influence of CYP2B6 (the drug metabolizing enzyme primarily responsible for EFV metabolism) genotype. For example, those with the extensive genotype had median EFV concentrations < 1mg/L both during RIF-including TB therapy and after TB therapy; the proportion with subtherapeutic concentrations appeared to be greater during TB therapy. Those with the slow genotype, however, had 3-fold higher EFV concentrations compared with the extensive genotype. The emerging consideration is the presence of isoniazid (INH), which is an inhibitor of CYP2A6, a minor pathway of EFV metabolism. Those individuals with the slow 2B6 genotype had 35% higher EFV concentrations in the presence of RIF and INH. This might arise because of INH inhibition of this minor pathway of EFV clearance.
This study illustrates how complex management of drug interactions can be; the results show an interplay among dose, body weight, drug metabolizing genotype, and drugs with competing effects of induction and inhibition. In both children and adults, the only way to sort out these effects would be to actually measure drug concentrations (in this case of EFV). If clinicians aren't going to measure EFV concentrations, and this is not a routine practice, I am inclined to still recommend an increase in the EFV dose if given with RIF. The most likely downside of a dose increase is EFV-associated CNS adverse effects, which if it occurs indicates the EFV concentrations are too high and that the increased dose isn't necessary. In my opinion, this scenario is preferable to virologic failure because of too low of EFV concentrations (when given with RIF), which by the time recognized with an increase in plasma HIV RNA, can't be corrected with an EFV dose increase.
Elvitegravir+Cobicistat drug interactions with rosuvastatin, atazanavir and rifabutin: OK, Not Yet and Not Recommended.
Drug-drug interactions between EVG+COBI and rosuvastatin, atazanavir and rifabutin were evaluated in 3 cohorts of healthy volunteers. The rosuvastatin dose was 10 mg and that for EVG+COBI was 150/150mg. The ATV dose was 300 mg, but the EVG+COBI doses were 85/150mg. The reduced EVG dose is because EVG is metabolized by glucuronidation and ATV is an inhibitor of glucuronidation. The RBT dose was 150 mg every other day when given with EVG+COBI 150/150mg.

Administration of EVG+COBI and rosuvastatin at the usual starting dose of 10 mg/day looks fine. Because rosuvastatin concentrations are increased, however, clinicians should monitor for adverse effects, primarily rhabdomyolysis and liver enzyme elevations especially with use of higher rosuvastatin doses. I think there is work left to do prior to recommending the use of ATV with EVG+COBI. ATV trough concentrations are decreased 20% and the variability in trough concentrations is doubled, when compared with ATV/RTV. There are no data that I am aware of for the combination of ATV with EVG+COBI and + TDF, and the potential of the addition of TDF to decrease ATV trough concentrations even further. So, my conclusion is not yet to an ATV with EVG+COBI combination. Finally, RBT substantially decreases both EVG and COBI concentrations, and concomitant administration is not recommended; this recommendation will also apply to rifampin and rifapentine, thus EVG+COBI will not be an option for the HIV-TB coinfected person requiring rifamycin therapy.
Dolutegravir and Rifabutin: Co-administration Looks to be OK.
Poster P_11 described the PK of dolutegravir, 50 mg, when given alone and with rifabutin, 300 mg once daily, for 14 days to healthy volunteers. The geometric mean ratios for the PK parameters of DTG (DTG + RBT to DTG) were: AUC, 0.95; Cmax, 1.15; and Ctrough, 0.70. These data indicate no effect of RBT on the AUC and Cmax of DTG, but a 30% reduction in the trough concentrations of DTG. The authors concluded that based on the PKPD relationships that have been described for DTG, this reduction is unlikely to be clinically significant. I agree. The published PKPD relationship (seehttp://www.natap.org/2011/HIV/090511_03.htm ) and the most recent 96-week results from the SPRING clinical study (see http://www.natap.org/2012/CROI/croi_22.htm) provide support for this conclusion. This is actually a very good example of the importance of a well-described clinical PKPD relationship - because it provides a scientific basis to predict whether a given reduction in concentrations is likely to be associated with a loss of response. I think it is warranted to confirm that adequate DTG concentrations are achieved in HIV and TB co-infected persons, and to obtain information on RBT concentrations as well.
As a reminder, a related drug interactions study between DTG and rifampin (RIF) was presented at CROI 2012 (see http://www.natap.org/2012/CROI/croi_46.htm ). This study found that a double dose of DTG, 50 mg twice daily, when given with RIF achieved DTG concentrations similar to 50 mg once daily without RIF.
For DTG with either RIF or RBT, clinical validation of safety and efficacy is necessary. That said, these results look promising as much-needed therapeutic options for treatment of HIV and TB coinfection.
IV. Clinical Pharmacology of Hepatitis C Therapy and Drug-Drug Interactions
First an update from CROI and late-breaker data presented (Abstract 771LB) on drug-drug interactions studies with the HCV protease inhibitor boceprevir (BOC) when given with RTV-boosted ATV, LPV or DRV. This interaction study found that:
· BOC reduces the concentrations of all PIs from 34% to 44%;
· BOC reduces the concentration of RTV; and
· LPV and DRV decrease BOC concentrations by 32% to 45%, while ATV does not effect BOC concentrations.
On the basis of these data, a Dear Health Care Provider letter of February 6, 2012 from Merck Inc. recommended that BOC not be coadministered with these PIs. Since CROI, the FDA has now weighed in, with a warning as well to not coadminister (see FDA http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm291144.htm ).
The interactions between BOC and the RTV-boosted PIs illustrate the current drug interaction challenges of managing concomitant therapy with ARVs and the new HCV direct acting antivirals. At this conference, Dr. Jennifer Kiser from the University of Colorado gave an outstanding presentation on understanding and managing these interactions. One of the most useful take home points from Dr. Kiser's talk was a summary of our ability to predict these interactions. She described interactions that were consistent with predictions, such as the reduction in HCV PI concentrations associated with the known induction by EFV of CYP3A. She described some interactions as not quite consistent with what we thought would happen, but not complete surprises, such as the prediction that neither BOC nor TVR would affect tenofovir (TFV) concentrations, yet both actually do increase TFV. And, finally, the real surprises, most notably that while we would expect RTV to increase BOC and TVR concentrations through an inhibition of CYP3A, but, the concentrations of the HCV PIs are actually reduced in the presence of RTV. For clinicians, I think the message is: we are at or beyond our ability to confidently predict interactions (two-, three-way or more) among ARVs and HCV PIs, which are all substrates, inhibitors and inducers of drug metabolizing enzymes and membrane transporters. If there are not PK data that provides support for using these combinations, I would not recommend it.
To drive the point home about predictable and unexpected interactions, here are summaries of two studies presented at the conference.
Telaprevir with Etravirine or Rilpivirine: OK and I would be careful.
TVR is an substrate and inhibitor of CYP3A and ETR is a substrate and weak inducer of CYP3A. On this basis, you would predict that significant interactions between these 2 drugs would be unlikely. And, that is what was shown (Abstract O-18) in a healthy volunteer study. There was no change in ETR PK parameters with coadministration of TVR; there was a 16% reduction in the AUC and a 25% reduction in Ctrough of TVR when given with ETR. This magnitude of reduction in TVR concentrations seems unlikely to adversely affect HCV response so coadministration should be OK, but this should be confirmed in patients.
Rilpivirine (RPV) is a CYP3A substrate and given that TVR is an inhibitor of CYP3A, you might expect that RPV concentrations would be increased with coadministration. And, indeed that is what was seen in this healthy volunteer study (also Abstract O-18). First, TVR concentrations just slightly reduced (8% reduction in AUC) when combined with RPV. RPV concentrations were increased in the presence of TVR: AUC, 79% increase; Cmax, 47%; and Ctrough, 89%. In healthy volunteers, 75 mg and 300 mg doses (3-fold and 12-fold) above the recommended dose of 25 mg once daily were shown to cause cardiac conduction abnormalities (prolongation of the QTc interval). Thus the recommendation for the lower dose of RPV and that it should be used in caution in persons with a known risk of Torsade de Pointes. The authors concluded this increase in RPV concentrations was not clinically relevant for increased QTc prolongation. Based on the data available, I am not quite ready to go along. We don't know what the interpatient variability was for the increase in RPV concentrations; the 90% confidence interval for the increase in RPV Ctrough was 1.5 to 2.35 indicating some patients had increases greater than 2-fold. I believe, given then the interpatient variability in RPV concentrations, that some patients taking the combination of TVR and RPV may reach RPV concentrations in the range of those seen with the 75 mg dose. Once again, I reiterate the need for confirmation of this interaction in patients. I would also recommend this combination be used very cautiously, and should probably not be used in patients receiving any other drugs known to increase RPV concentrations, in patients with refractory hypokalemia or hypomagnesemia, or with drugs known to prolong the QTc interval or that are a risk for Torsade de Pointes.
Boceprevir and Etravirine: co-administration not recommended at this time.
The results of a drug interaction study of BOC with ETR in healthy volunteers was reported in Abstract O_15. The predicted effects of this combination were that BOC concentrations would be decreased by ETR (as ETR is an inducer of CYP3A) and that ETR concentrations would be increased by BOC (because it inhibits CYP3A). But, just the opposite effects were actually seen. The AUC and Cmax of BOC were increased by 10%, while the Ctrough was decreased by 12%. ETR concentrations were decreased by BOC: AUC, 23% decrease; Cmax, 24%; and Ctrough, 29%. There was also greater interpatient variability in ETR concentrations when given with BOC. This study was done by investigators at the U of Colorado. They concluded the reduction in ETR concentrations could reach the boundary of clinical relevance. The investigators are conducting additional work to understand the mechanism(s) for this interaction, and potentially to assess whether it might be a result of protein binding displacement (requires measurement of unbound concentrations). Until additional information is known, I would recommend clinicians avoid combined use of BOC and ETR.
|
| |
|
|
|
|
|