 |
|
|
| |
Summary from AASLD 2013 for Hepatitis C
Washington 1-5 November 2013
|
| |
| |
Is there the right DAA combination for every challenging patient population?
Jurgen K. Rockstroh M.D., Professor of Medicine
University of Bonn, Germany
Correspondence:
Prof. Dr. J.K. Rockstroh
Department of Medicine I
University of Bonn
Sigmund-Freud-Str. 25
53105 Bonn
Germany
Tel.: +49-228-287 16558
FAX: +49-228-287 15034
e-mail: juergen.rockstroh@ukb.uni-bonn.de
Introduction
Although the introduction of the first DAA based triple therapy (telaprevir or boceprevir in combination with IFN/RBV) has already considerably improved outcome of HCV therapy and changed HCV treatment paradigms for HCV therapy, it became rapidly clear that patient populations exist which will remain difficult to treat including previous non-responders, particularly with concomitant cirrhosis, as well as patients pre and post liver transplantation with HCV recurrence. Also genotype 3 patients have emerged as a new difficult to treat patient group with IFN-free HCV regimens, again particularly in patients with cirrhosis. Moreover, with sofosbuvir achieving positive FDA panel approval and simeprevir just being approved in the USA and Canada at the end of November, the question remains how these drugs will be best used in the future after final approval in the next months. Will combinations of DAA from different companies on the basis of promising results from early pilot trials be sufficient to warrant their use outside of clinical trials? Will the various DAA combinations from a number of different companies be able to offer interferon-free HCV treatment regimens for all even for cirrhotic genotype 3 patients? This AASLD was able to create a great buzz around the fast moving and exciting HCV treatment story, which attracted over 10.000 delegates to attend AASLD in Washington and to experience these new advances in an unparalleled medicine success story. With a constantly increasing number of DAAs in development from conference to conference it becomes increasingly difficult to foresee where and in whom the individual drugs will be used eventually. Clearly pricing of the new drugs will also impact the use of the respective drug accordingly. The conference closed with a hepatitis C debrief by Mark Sulkowski which was simply excellent and everyone interested in HCV drug development should be encouraged to listen in into Marks wonderful summary through the AASLD website (1). The following conference report aims at covering the main HCV DAA trials and clinical relevant HCV management issues presented at AASLD in Washington from 1-5 November 2013.
Ending the silent epidemic
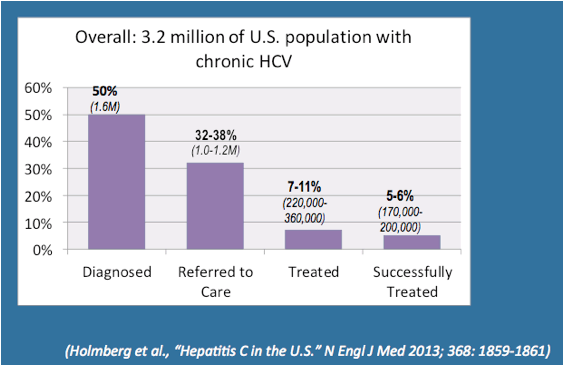
In an introduction to the hepatitis debrief towards the end of the conference Ronald Valdiserri summarized the best way forward to end the the HCV epidemic (2). He emphasized that improved HCV treatment outcomes will require timely diagnosis and linkage to care. He reported that during 2005-2011 at 8 US surveillance sites 217,755 persons were identified as newly HCV positive, whereby 49% lacked HCV-RNA testing (3). In a phone survey of 53,896 minority adults from 28 communities only 19% reported HCV testing despite 60% having a risk factor for HCV transmission (4). Of those tested 8,3% were HCV positive but only 44% were subsequently followed by a doctor. Readdressing the American HCV treatment cascade he highlighted that of the 3,2 Mllion Americans believed to have HCV only 50% (1,6 Million) know their diagnosis. 32-38% of these patients are presumed to be referred to care with only 7-11% being treated and eventually only 5-6% (170,000 - 200,000) being cured from their HCV infection (5). The corresponding American HCV treatment cascade he presented is depicted in Figure 1. Clearly these findings highlight that improved treatments with high cure rates also need to be implemented in a sector of medicine where up to now acess to diagnostics and subsequent linkage to hepatitis mangement have been more than challenging.
Figure 1: U.S, HCV Treatment Cascade (5)
Efficacy and safety of boceprevir and telaprevir: the real-life experience
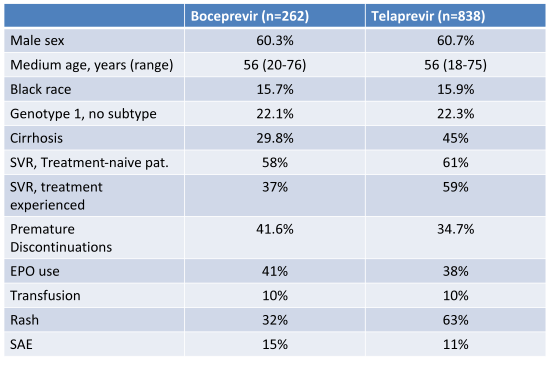
HCV-TARGET is an ongoing longitudinal observational study at 44 academic and 59 community centers (6). Demographic, clinical and virologic data and adverse events are collected throughout treatment and follow-up on sequentially enrolled patients, together with information on adherence to treatment futility rules at key time points. 61% of the included patients are already treatment experienced. 137/678 treatment experienced patients showed prior null response, most patients were relapsers (302/678). Virological results and baseline characteristics of the cohort are summarized in table 1. Indeed, overall response rates in this real-life cohort become lower with prior null response. However cure rates in the treatment naïve patients are reassuringly high. With regard to safety there appears to be a higher rate of SAEs than seen on the registrational trials but overall manageable.
Table 1: Experience with boceprevir or telaprevir + PEG-IFN/RBV in an observational cohort at academic and community sites: HCV-TARGET
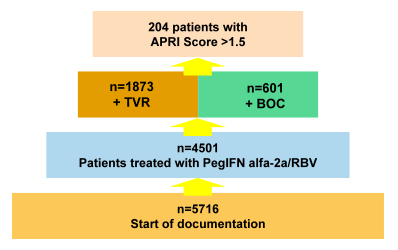
Data from the French CUPIC study (7) and an Austrian cohort (8) have previously demonstrated that patients with F3 or F4 stage fibrosis had a modest treatment outcome and a high rate of severe events, particularly in patients with low platelet counts <100,000/mm3 and serum albumin <35 g/L. In a German cohort, initiated by the Association of German Gastroenterologists in Private Practice (bng) and Roche, the impact of these baseline factors on treatment efficacy was analysed in this noninterventional "PAN" cohort (9). Only patients infected with HCV genotype 1, initiating treatment with BOC or TVR from October 2011 to March 2012 with an APRI score >1.5 were included into this analyses (see Figure 2).
Figure 2: Study design of the PAN cohort.
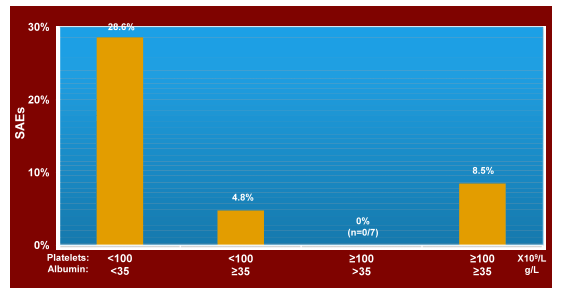
A total of 204 patients were analysed, 45 with boceprevir (BOC) and 159 with telaprevir (TVR) triple therapy. 47.1% of patients had liver cirrhosis (at least one result of sonography, histology, elastography or clinical appearance). 56.4% male, mean age 53.6 years, BMI 27.1 kg/m2. 65.2% of patients had ALT >3xULN, 33.3% platelets <100,000/μl and 17.1% albumin <35 g/L. Genotype-subtypes distribution was 1a 29.9%, 1b 52.0%, unknown/other 18.1%; High viral load (HCV-RNA>400,000 IU/ml) 74,4%. 147/204 patients (72.1%) were pretreated with 61 relapsers, 86 non-responders. Treatment discontinuation rate increased over time with 33.3% discontinuing before week 48. Preliminary data of 88 patients having reached an observational period of 60 of 72 weeks reveal 65.9 % treatment discontinuation and 25.0% SVR. The risk of SAE increased to 28.6% with platelet count <100,000/mm3 and serum albumin <35g/L (see Figure 3). These findings are clinically extremely relevant as they underline that patients with low albumin and platelet levels indeed are high risk for SAE development. It also suggests that outside of clinical trials far more patients are discontinued under first wave HCV-protease inhibitor based therapy.
Figure 3: Real World: Significant Adverse Events (SAEs)
Treatment in special patient populations: HIV/HCV coinfection
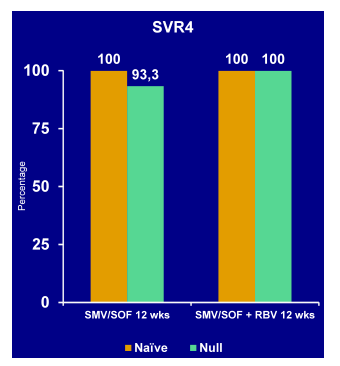
At AASLD various trials were reported from DAA-based therapy in HIV/HCV coinfected individuals which included data from trials with telaprevir and boceprevir in more real-life treatment scenarios including considerable patient numbers with more advanced fibrosis as well as previous treatment failure to dual therapy (10, 11). In addition SVR4 results from the phase III trial investigating the second-wave HCV protease inhibitor faldaprevir in combination with PEG-IFN/RBV (12) as well as the results from the first IFN-free regimen with Sofosbuvir and ribavirin (PHOTON trial) were reported (13).
Studies with telaprevir or boceprevir in HIV-coinfection
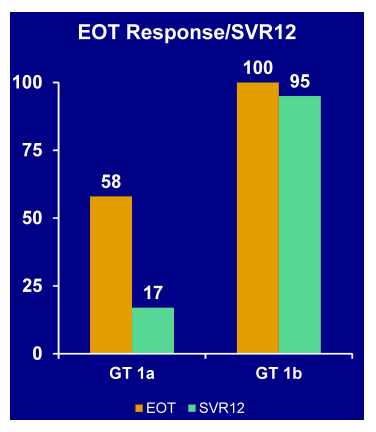
In the hepatitis poster session an update on two ANRS studies were presented which looked at the efficacy and safety of triple HCV therapy (with either telaprevir or boceprevir) in previous non-responders to dual PEG-IFN/RBV therapy (10,11). In the two trials patients with previous null-response and cirrhosis however, were excluded because of the overall low probability of treatment response. At CROI interim results from week 16 had been presented, here at AASLD 48 week results were reported for the first time (14,15). In the telaprevir ANRS HC26 study overall 69 patients who received at least one dosage were included (10). Background HIV therapy contained ATV, ATV/r, EFV, RAL, TDF, FTC, or 3TC. The study design is depicted below (Figure 4):
Figure 4: Study design of the ANRS HC26 TelapreVIH Study.
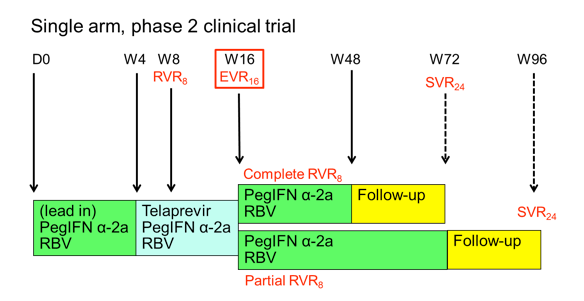
Patients started with a 4 week lead-in of PEG-IFN/RBV. This approach, which is outside the labelling of telaprevir, was chosen to be able to compare the results to the study design wise similar boceprevir study and because data from telaprevir studies in HCV mono-infection in treatment experienced patients had potentially promised some benefit for this approach. After week 4 telaprevir was added for 12 weeks. Patients who achieved a complete rapid virological response at week 8 (RVR8) defined as HCV-RNA <15 IU/mL received 32 weeks of PEG-IFN/RBV after stopping telaprevir at week 16 after 12 weeks of triple therapy (full treatment: 48 weeks). Whereas patients who only obtained a partial RVR8 (15 IU/mL < HCV-RNA < 1000 IU/mL) received an additional 56 weeks of PEG-IFN/RBV (full treatment: 72 weeks). Telaprevir was administered as 750 mg q8h (1125 mg q8h with Efavirenz) and Peg-IFN α-2a as 180 μg sc / week. Ribavirin was dosed as 1000 mg / day (≤75 kg) and 1200 mg /day (>75 kg). EPO, G-CSF, and TPO-R agonists were allowed within the study. Futility rules for telaprevir were HCV-RNA > 1000 IU/mL at week 8 or week 12 or virological breakthrough at any time. For study inclusion patients needed to have CD4 ≥ 200 cells/mm³ and ≥ 15%, as well as plasma HIV-RNA levels < 50 copies/mL. At baseline median CD4-count was 630 cells/mm³ (range 459-736) and HIV-RNA was below 50 copies in 99% of patients. Interestingly, 11 (18%) patients had F3 fibrosis and 16 (23%) F4 fibrosis upon inclusion to the study, allowing studying safety and efficacy of telaprevir based triple therapy in this more difficult-to-treat patient population. 39% of patients were previous relapsers to dual therapy, 9% had previous viral breakthrough, 22% were partial responders and 30% null-responders. The week 48 efficacy results in an ITT-analysis are presented below in Figure 5.:
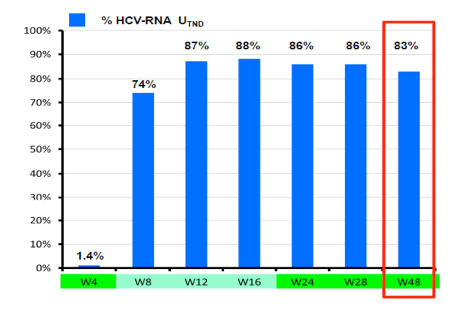
Figure 5: Results: Telaprevir + PegIFN + RBV in HIV/HCV co-infected patients with previous virologic failure on IFN +RBV: virologic response at week 48 (ITT, n=69)
UTND= target not detected
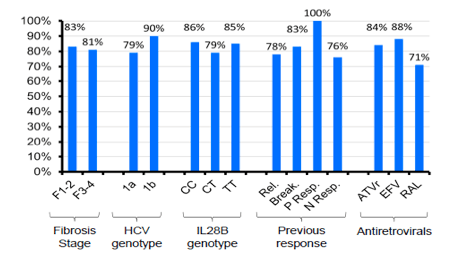
With 83% being HCV-RNA negative at week 48 in this patient population with 30% previous null-responders and 40% F3/F4 fibrosis this study showed surprising high efficacy results at EOT for most patients. Most interestingly efficacy remained high throughout all sub analyses and was independent of prior fibrosis stage, previous HCV treatment response or background ART (see Figure 6). With regard to safety 20% of patients were reported to develop grade 4 adverse events. Most common grade 4 adverse events were haematological. 65% of patients received EPO, 23% blood transfusions. So although side effects were manageable complications under triple therapy with telaprevir in this more challenging patient population were frequent.
Figure 6: Virological response by baseline characteristic
The second ANRS study looked at the efficacy and safety of a boceprevir containing triple therapy again in 64 previous IFN/RBV non-responders (11,15). The study design is depicted below in Figure 7:
Figure 7: Study design: Multi-center single arm open label Phase 2 Trial
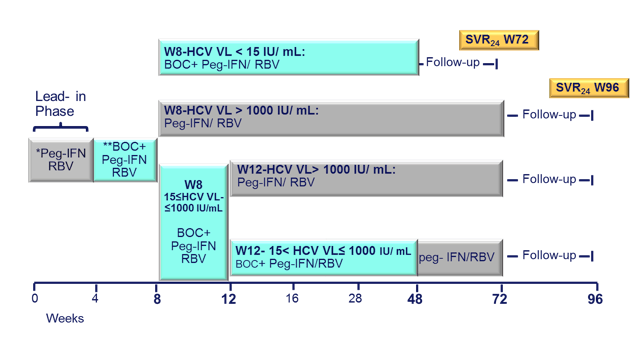
Following a lead-in with dual therapy boceprevir was added at week 4. Complex futility rules were in place during the study warranting discontinuation of boceprevir in all patients with a HCV viral load above 1000 IU/ml at week 8 as well as in week 12. All HCV drugs were discontinued if HCV viral load was >1000 IU/ml at week 16 or still detectable at week 28 and in case of virological breakthrough. Duration of therapy was dependent on initial response as shown above. Patients who achieved a rapid virological response and had an HCV-RNA <15 IU/ml at week 8 were treated for 48 weeks, patients with slower decline in HCV RNA were treated for 72 weeks. Patients recruited for the trial had to be on stable ART for at least 3 months, with at least three molecules among ATV (rtv boosted or not), RAL, TDF, ABC, FTC, or 3TC. Again only previous non-responders to dual therapy with HCV genotype 1 infection were included. Null-responders with cirrhosis were excluded from this trial. Overall, 17% of patients at baseline had F4 fibrosis, 33% were previous null-responders. Only patients who received ≥ 1 dose of treatment Interim week were included into this first interim week 16 efficacy analyses. The corresponding response rate (Patients (%) with HCV-RNA < 15UI/ mLs ) at 48 weeks are depicted below (Figure 8):
Figure 8: HCV- Virological response at week 48: Patients (%) with HCV-RNA < 15UI/ mL
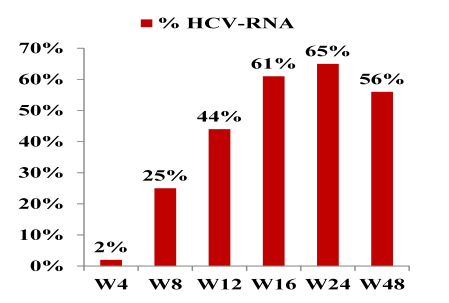
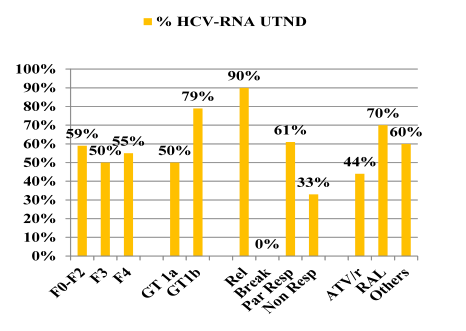
Overall 56% achieved undetectability at week 48. Response rates at week 48 were best for previous relapsers (with 90% < 15 IU/ml) versus patients with previous partial response (61% < 15 IU/ml) and null-responders (33% < 15 IU/ml), respectively. Notably, none of the previous breakthroughs responded raising the question whether this group of patients may be more difficult to treat (patient numbers are very small though). No difference in response was seen according to fibrosis at baseline. Figure 9 summarizes the results of the corresponding subanalyses.
Figure 9: Virological response according to baseline characteristics
In summary, these first two studies in previous non-responders to dual therapy with PEG-IFN/RBV and a considerable proportion of patients with more advanced fibrosis stages demonstrates very high on treatment response rates in this more challenging to treat patient population. Overall, it appears as if boceprevir works less well in the context of previous non-responders or breakthroughs. This should remind us not to postpone HCV therapy for all in hope of easier and less toxic HCV treatment regimens in the future but also to at least consider the improved treatment options we already have in our hands which preferably should be used in those patients where risk for development of HCC or progression to cirrhosis and eventually hepatic decompensation is a real threat. Cure rates are particularly promising in treatment-naïve and relapser patients. Safety appears manageable but haematological adverse events are common.
More data on HCV combination therapy with telaprevir was reported from the INSIGHT trial. This study evaluated the efficacy and safety of response guided telaprevir/PEG-IFN/RBV therapy in HCV treatment naïve and experienced patients (16). In the study patients who were on stable, suppressive HIV antiretroviral therapy (ARV) including atazanavir (ATZ)/ritonavir (r), efavirenz (EFV), darunavir (DRV)/r, raltegravir (RAL), etravirine (ETR) or rilpivirine received TVR 750mg q8h (1125mg q8h if on EFV) plus PEG-IFN (180μg once-weekly) and ribavirin (800mg/day) for 12 weeks, followed by an additional 12 weeks (HCV treatment-naïve and relapse patients with extended rapid viral response [eRVR]) or 36 weeks (all others) of PEG-IFN/RBV alone. Interim 12-week results in 162 patients were reported. At baseline 30% of patients had bridging fibrosis or cirrhosis. 64 patients were treatment-naïve, 29 prior relapsers, 18 prior partial responders, and 51 prior non-responders. The proportion of patients with undetectable HCV-RNA at week 12 was high with 72%, with almost half of the patients (49%) achieving eRVR. Again promising early response rates were obtained, but due to the short follow-up further conclusions cannot be drawn at this time-point.
Studies with new DAAs in HIV/HCV coinfection
The STARTVerso4 (SV4) study was set up to assess efficacy and safety of the second wave HCV protease inhibitor faldaprevir (FDV) plus PEG-IFN/RBV and evaluate a 24-week, shortened treatment duration in HIV patients coinfected with chronic HCV genotype (GT) 1 (12). SV4 is an open-label, sponsor-blinded study in HCV/HIV coinfected patients who were HCV treatment-naïve (TN) or relapsed after previous HCV therapy. In arm A patients received FDV 120 mg QD and PEG-IFN/RBV for 24 weeks; in arm B: patients received FDV 240 mg QD plus PEG-IFN/RBV for 12 weeks and were randomized at week12 to receive a further 12 weeks of FDV/PEG-IFN/RBV or PEG-IFN/RBV alone (see Figure 10). At week 24 all patients who achieved early treatment success (ETS, HCV RNA <25 IU/mL detected or undetected at W4 and undetected at W8) were re-randomized 1:1 to stop treatment at week 24 or continue PEG-IFN/RBV up to W48. Patients without ETS received PEG-IFN/RBV through 48 weeks. In order to correct for drug-drug interactions patients on HIV protease-based antiretroviral therapy (ART) or efavirenz were allocated to receive FDV 120 mg or 240 mg QD, respectively; those receiving other allowed ART (raltegravir or maraviroc) or no ART were randomized to either FDV dose. The primary endpoint was SVR12. In the AASLD interim analysis SVR4 rates were presented.
Figure 10: Study design of the STARTVerso4 Phase III open-label, sponsor-blinded study in treatment-naïve and relapser patients with chronic HCV GT-1 and HIV infection
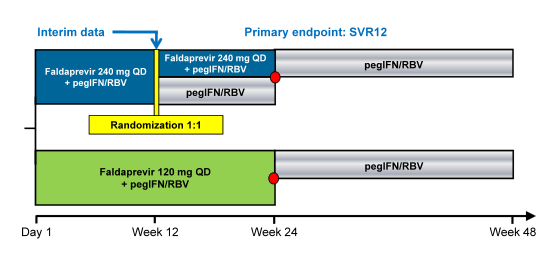
The red dot resembles patients with HCV RNA below LLoQ, at Week 4, and HCV RNA below LLoQ target not detected at Week 8 (=ETS). In total, 308 patients were treated (mean age 47 years, 81% male, 83% Caucasian, 14% Black/African American, 29% ≥F3 liver fibrosis, 79% GT1a, 66% IL28B non-CC rs12979860). ART was used by 96% of patients. A successful SVR 4 response was achieved by 220/308 (74%) of the total patient population being even higher in previous relapsers with 60/69 (87%) (Please see Figure xx). Most common AEs were nausea (37%), fatigue (34%), and diarrhea (27%). Study medication was discontinued due to AEs in 7% of patients in Arm A and 8% in Arm B. Serious AEs occurred in 14% and 8% of patients, respectively. Hemoglobin ≤8.5 g/dL occurred in 6% of patients.
Figure 11: SVR rates in Startvers4
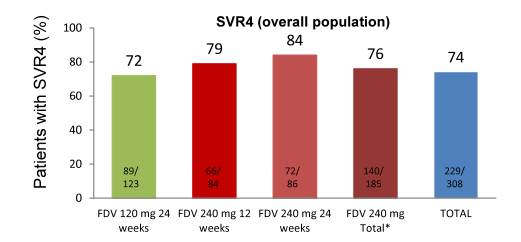
Overall, virological breakthrough was observed in 8% of patients, relapse occurred in 4% of patients. No difference in SVR4 rate was seen between GT 1a and 1b. Also no difference in outcome was observed by baseline cirrhosis (see Figure 12) or ART. Even more importantly, 80% of patients achieved early treatment success which qualified them for being randomized to shorter (24 weeks) or longer treatment duration (48 weeks). Interestingly, no difference in outcome was obtained for ETS patients irrespective of treatment duration or faldaprevir dose. Overall, this once daily second wave HCV protease inhibitors promises good efficacy for most patients with short treatment duration following response guided therapy. This is the first HCV PI which can be administered concomitantly with boosted darunavir.
Figure 12: SVR4 rate by baseline cirrhosis
Clearly with regard to coinfection studies the results from the first interferon-free trial in coinfection were the most exciting study results presented at AASLD. The so called PHOTON 1 study evaluated the safety and efficacy of the oral HCV NS5B inhibitor sofosbuvir (SOF) with ribavirin (RBV) in HIV -seropositive patients coinfected with HCV genotypes 1, 2 or 3. Patients included into the study received SOF 400 mg QD and RBV 1000-1200mg/day; GT 1 patients received 24 weeks and GT 2/3 patients received 12 weeks of treatment. The study design is shown below in Figure 13. At the AASLD meeting only results from the treatment-naïve arms were reported.
Figure 13: Study design of the PHOTON-1 trial.

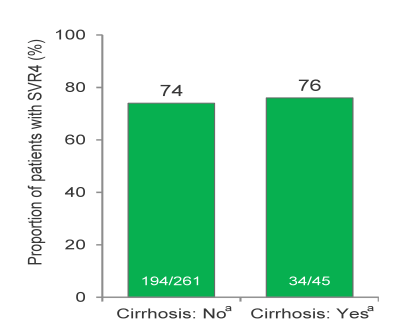
Overall, 4% of the treatment -naïve GT1 patients (n=114), 4% of the GT2 patients (n=26) and 14% of the GT 3 patients (n=42), respectively had cirrhosis at baseline. 93-98% of patients were on ART and had stable HIV disease. Among patients with GT1, GT2 and GT3, SVR12 was achieved in 76% (87/114), 88% (23/26) and 67% (28/42), respectively (see Figure 14). Among the 27 GT 1 patients without SVR12, viral relapse was confirmed in 25 patients. In contrast no relapse occurred in the GT 2 patients but in 12 GT 3 patients without SVR12. Therefore it appears that treatment duration of 12 weeks is sufficient for GT2 treatment whereas for GT3 potentially longer treatment durations are required to prevent relapse (currently being investigated in PHOTON-2). For GT1 the majority of patients respond well to an interferon-free regimen of sofosbuvir and ribavirin but the high relapse rate suggests that there exists a subset of patients which will require DAA combination therapy. Overall, these results demonstrate that interferon-free treatment strategies are a very close reality for the majority of patients. As response rates are virtually superimposable to findings in monoinfected patients these findings also underline that probably HIV patients no longer should be treated as an independent patient population (other than for their potential for drug-drug interactions between HCV drugs and antiretroviral therapy) but on average can be treated like HCV mono-infected subjects.
Figure 14: On-treatment and Sustained Virologic Response
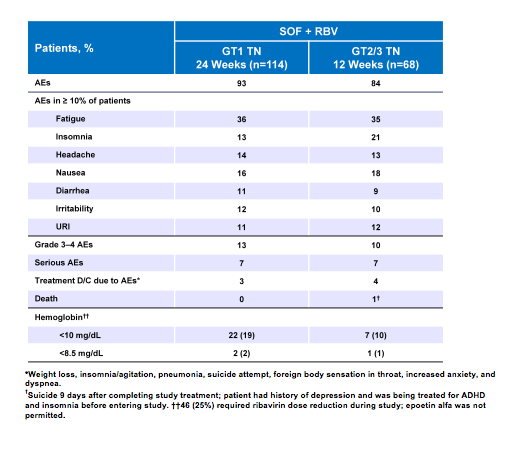
Treatment discontinuations due to adverse events (AEs) were uncommon (3-4%) in the PHOTON trial and grade 3/4 AEs were reported in only 13% of the treatment-naïve GT1patients and 10% in the treatment-naïve GT2/3 patients. Most adverse events were typical side effects under ribavirin based therapy (see table 2).
Table 2: Safety findings from the PHOTON-1 trial
Patients pre- and post-liver transplantation
Patients undergoing liver transplantation because of HCV-associated advanced liver disease almost all develop HCV recurrence following transplantation. In the setting of immunosuppression to prevent organ rejection HCV recurrence often takes an unfavorable course and can rapidly lead to advanced fibrosis. In addition treatment outcome of dual therapy with PEG-IFN/RBV in the pre or post-transplant setting has been associated with poor outcomes. Unfortunately, this has had a negative impact on overall survival after transplantation in comparison to transplantation for other reasons. Therefore, new and better tolerated HCV treatment options are eagerly awaited for this challenging to-treat patient group.
At AASLD results from a Phase 2, open-label study of SOF + RBV for up to 48 weeks in patients with HCV listed for liver transplant for hepatocellular carcinoma (HCC) were presented (20). The study design is depicted below (Figure 15):
Figure 15: Study design of pretransplant sofosbuvir and ribavirin to prevent recurrence of
HCV infection after liver transplantation

In this open-label study, patients with chronic HCV infection of any genotype (GT) listed for liver transplantation for hepatocellular carcinoma (HCC) received up to 48 weeks of sofosbuvir 400 mg/day and ribavirin 1000-1200 mg/day before transplantation. All patients had HCC within Milan criteria and well compensated cirrhosis (Child-Pugh-Turcotte score of ≤7). The primary endpoint was virologic response (HCV RNA <25 IU/mL) 12 weeks after LT in patients who had HCVRNA <25 IU/mL at their last measurement prior to LT (SVR12). Post-LT immunosuppressive regimen was tacrolimus plus prednisone with or without mycophenolate mofetil. A total of 61 patients (24 GT 1a, 21 GT 1b, 8 GT 2, 7 GT 3, and 1 GT 4) were enrolled and received study drug (mean age 59 years, 80% male, 10% black, 20% Hispanic, 75% previously treated, with a mean baseline HCV RNA of 6.14 log10 IU/mL [range 4.06-7.23 log10 IU/mL]). To date, 44 patients have undergone LT. Of these, 41 were
<25 IU/mL and 3 were >25 IU/mL at last HCV RNA measurement prior to liver transplantation. The post virological response rates are summarized in Figure 16:
Figure 16: Post transplantation virological response rates
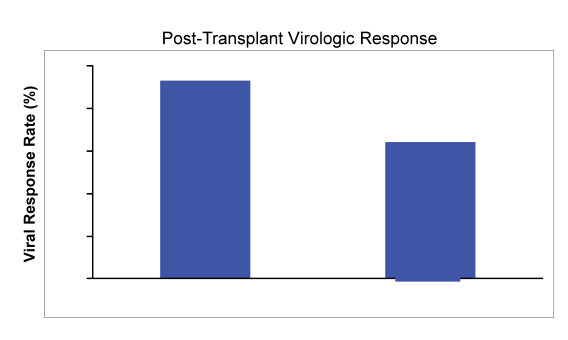
Overall, sofosbuvir and ribavirin therapy was safe and effective in patients with well compensated cirrhosis, and prevented post-transplant HCV recurrence in 64% of patients who had HCV RNA < 25 IU/mL prior to transplant. Of the 44 transplanted patients 3 subjects were >LLOQ at transplant, 1 subject has not reached pTVR12, and 1 subject was lost to follow-up at Week 8 post-.transplant. Eleven patients had recurrence, and 3 (5%) died related to transplantation-associated complications. Most interestingly the number of consecutive days with HCV RNA target not detected prior to transplant appeared to be the strongest predictor of post-transplant HCV recurrence. The importance of time being undetectable is nicely summarized in Figure 17 below:
Figure 17: Days Continuously target not detected (TND) Prior to Transplant: Post transplant virological response versus. recurrence in GT 1-4
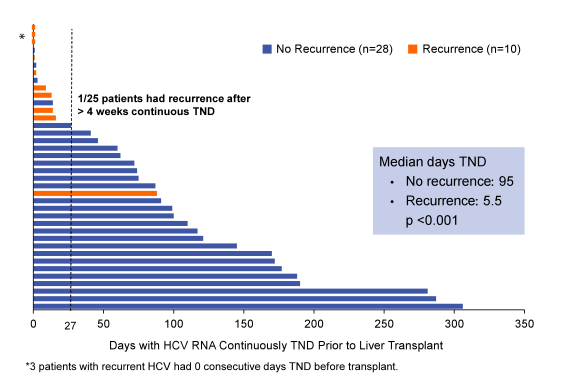
In multivariate analyses only days being target non-detected remained significantly associated with risk for post-transplant HCV recurrence whereas IL28b genotype and genotype 1 subtype revealed no significant impact in the analyses. Clearly, these findings imply if the organ transplantation occurs just after having reached target not detected this may not be sufficient to prevent HCV recurrence. However, if the patient has already been undetectable for quite some time post-transplant recurrence can be prevented efficiently.
Another exciting study in the context of liver transplantation was the presentation of a prospective, multicenter, open-label, IFN-free pilot study of SOF + RBV for up to
24 weeks in naïve and treatment-experienced patients undergoing liver transplantation with recurrent HCV infection (21). The study design of this pilot trial is depicted in Figure 18:
Figure 18: Phase 2 Post-Liver Transplant Study

In this ongoing single-arm, open-label interferon-free pilot study, naïve and treatment-experienced patients with recurrent HCV infection (any HCV genotype) after liver transplantation received up to 24 weeks of SOF 400 mg and RBV (dose regimen starting at 400 mg/day). Patients were ≥18 years of age and at least 6 months post-transplantation. Overall, 40 patients (22 GT 1a, 11 GT 1b, 6 GT 3, 1 GT 4) were enrolled into the study. Mean age was 59 years, 78% of patients were male, 8%black, 3% Hispanic: 88% previously treated [9 previously treated with protease inhibitors], mean baseline HCV RNA of 6.55 log10 IU/mL [range 4.49- 7.59 log10 IU/mL], and 40% cirrhosis. The mean years since liver transplantation were 4.3 years (range 1.02-10.6). The mean duration of treatment was 23.2 weeks (15.1-24.4 weeks). Five patients remain on treatment and 2 patients have discontinued treatment due to adverse events (AE) of pneumonia and HCC progression. By week 4 on treatment, 100% of patients had HCV RNA <25IU/mL. The interim SVR4 rates are shown in Figure 19:
Figure 19: SOF + RBV for Established Recurrent HCV Post-Liver Transplant: Virologic Response Interim Results
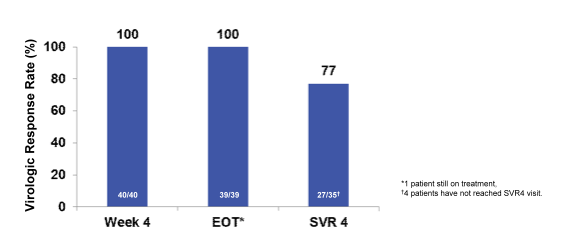
There were no episodes of acute or chronic rejection. No drug interaction dose adjustments of immunosuppression were required throughout the study. Treatment with SOF and RBV was well tolerated. Six patients experienced treatment-related serious adverse events not related to SOF and 31 patients (78%) experienced treatment-emergent, treatment-related AEs mostly related to ribavirin use. Seven subjects (18%) experienced mild to moderate anemia. The most common adverse events were fatigue, arthralgia, headache, and diarrhea. In conclusion administration of SOF and ribavirin was safe and effective. In summary, the high cure rates under this easy to take well tolerated regime are highly promising and open the way to much simpler treatment of HCV recurrence following liver transplantation.
Clearly interferon-free strategies are highly desirable in post OLTX patients due to the high risk for adverse events in this challenging patient population under interferon/ribavirin therapy. At this year AASLD numerous studies in post liver transplant patients were reported for telaprevir or boceprevir based triple therapy (22-24). The main virological outcome data as well as safety findings are summarized in table 3:
Table 3: Telaprevir or boceprevir plus PEG-IFN/RBV for treatment of recurrent HCV after liver transplantation.
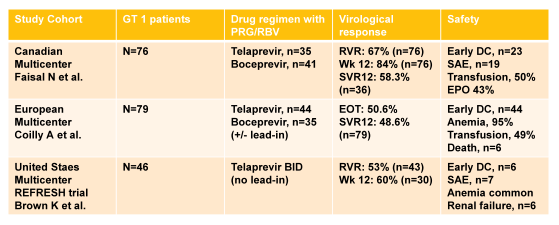
A triple therapy regimen including a PI for recurrent genotype 1 HCV in liver transplant recipients appears significantly more effective than treatment with P/R only, with projected SVR rates between 48 and 58%. Adverse events are however common, requiring careful monitoring and mostly intensive anemia management including EPO, transfusions and ribavirin dose reductions.
New DAAs in combination with PEG-IFN/RBV
At this year's AASLD mostly meta-analyses from the phase III programs for the soon (faldaprevir, sofosbuvir) or just (simeprevir: end of November 2013) recently licensed DAAs were presented.
QUEST-1 and QUEST-2 were Phase III, randomized, double-blind studies evaluating simeprevir (SMV), an investigational, once-daily (QD), oral HCV NS3/4 protease inhibitor, with PegIFN α-2a (QUEST-1) or PegIFN α-2a/2b (QUEST-2) plus RBV (PR) in treatment-naïve HCV genotype-1 (GT 1) adult patients. The pooled analysis presented at AASLD from these registrational trials focused on the comparison of the efficacy of SMV/PR versus PR in patient sub-groups (IL28B genotype, METAVIR score, HCV GT subtype, and baseline HCV non-structural protein 3 [NS3] Q80K polymorphism) (25). The pooled analysis included 785 patients: IL28B TT, 14.6%; METAVIR F4, 10.4%; GT 1a, 48.4%; Q80K+, 16.8% (all but 2 patients with Q80K had GT 1a). Overall, SVR12 rate was significantly superior with SMV/PR versus PBO/PR (80.4% versus 50.0%; p<0.001). Among patients who achieved rapid virologic response (RVR; defined as HCV RNA < 25 IU/mL undetectable at Week 4) in the SMV/PR arm (77.5% overall), 89.6% went on to achieve SVR12. The results for the main subgroups are shown below in Figure 20.
Figure 20: Pooled analysis of simeprevir plus PegIFN/RBV for treatment of treatment-naïve patients with genotype 1 infection versus PBO/PR: SVR12 rates
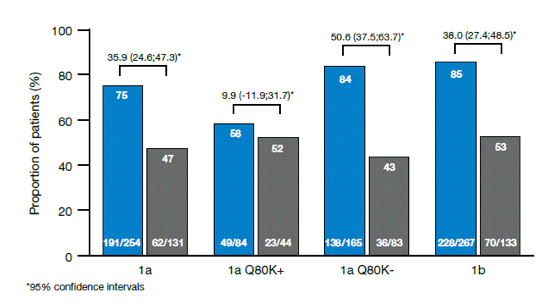
SVR12 rates were 94.7%, 92.3%, 85.4% and 83.9%, respectively in patients receiving SMV/PR who had CC IL28B genotype, baseline HCV RNA ≤ 800,000 IU/ml, GT 1b and METAVIR F0-F2. SVR12 rates were also significantly higher in the SMV/PR vs PBO/PR arm for each of the difficult-to-treat subgroups analyzed, including patients with cirrhosis, patients with TT IL28b genotype and patients with baseline Q80K. In conclusion simeprevir conferred clinical benefit across all patient sub-populations, including not only those with positive predictors of response (IL28B CC, METAVIR F0-F2, GT 1band GT 1a/Q80K-), but also patients with unfavorable baseline characteristics, such as IL28B TT, METAVIR F4 and GT 1a/Q80K+. Interestingly, week 4 HCV RNA determination allowed identification of those patients who are most likely to benefit from SMV therapy. Discontinuation rate because of adverse events was low with 3% in the pooled analyses.
A further simeprevir + PEG/IFN study called PROMISE was also presented in the poster session (26). This double-blind, multicenter, Phase III study evaluated simeprevir (SMV), plus PegIFNα-2a/RBV (PR) in HCV genotype 1 patients with prior relapse. Patients who had received IFN-based therapy for ≥24 weeks and relapsed ≤1 year thereafter were randomized 2:1 to SMV 150mg QD (12 weeks) plus PR (response guided 24/48 weeks) or to placebo (PBO, 12 wks) plus PR (48 weeks). The ITT population comprised 393 patients. SVR12 was significantly higher with SMV/PR versus PBO/PR overall (79.2% vs 36.8%; p<0.001). SVR12 was also statistically significantly higher with SMV/PR vs PBO/PR in difficult-to-cure IL28B TT genotype, METAVIR F4 and genotype 1a patients, and higher in genotype 1a patients with baseline Q80K (46.7% vs 27.8%).
Overall, simeprevir/PEG-IFN/RBV treatment promises easier HCV therapy than with telaprevir or boceprevir as simeprevir is dosed once daily. The safety also looks better than previous triple therapy. Concerns remain with regard to efficacy in patients with a baseline Q80K which was shown to be present in 13.7% of all HCV genotypes, but being found most frequently in genotype 1a patients (29.5%; highest in GT1a patients from North America 48.1%) (27).
STARTVerso(SV) 1 (Europe and Japan) and SV2 (North America, South Korea, and Taiwan) assessed faldaprevir (FDV), a potent once-daily HCV NS3/4A inhibitor plus pegylated interferon alfa-2a and ribavirin (PR) in treatment-naïve patients with chronic HCV genotype 1 (GT1) infection. Patients were randomized 1:2:2 to receive 48 weeks (W) of PR plus: placebo (Arm 1); FDV 120mg QD for 12W or24W (Arm 2, response guided in SV1); or FDV 240mg QD for 12W (Arm 3). Patients in Arms 2 and 3 stopped all treatment at W24 after achieving early treatment success (ETS, HCV RNA <25IU/mL detected or undetected at W4and undetected at W8). Here at AASLD the results from a pooled analysis of the two trials were presented. Figure 21 shows the SVR12 rates by region.
Figure 21: SVR12 rate by region from the pooled analyses from STARTVerso 1 an 2.
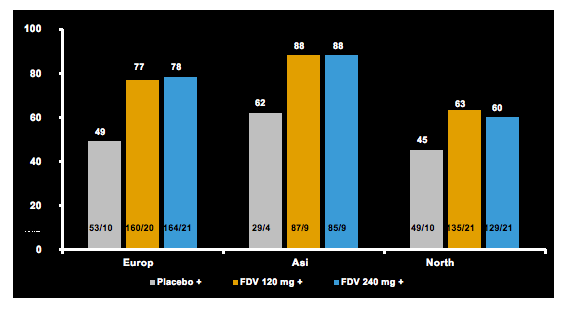
Results were similar between patients treated with FDV 120mg or 240mg in the whole population and across baseline subgroups. For Arms 1, 2, and 3, discontinuation of all study medication due to AEs was reported in 4%, 5%, and 8% of patients, respectively. Serious AEs occurred in 6%, 7%, and 8% of patient s. Severe rash was reported in 1% of patients who received either FDV or placebo; no severe photosensitivity reactions were observed (please keep in mind that sun-blockers were prophylactically applied in the study). Hemoglobin ≤8.5 g/dL occurred in 3%, 4%, and 3% of patients. Bilirubin >2.5 × the upper limit of normal occurred in 0.4%, 12%, and 46% of patients.
Overall, faldaprevir based triple therapy also allows for simplified therapy (faldaprevir is administered once daily) in comparison to telaprevir/boceprevir based HCV therapy with comparable high SVR rates.
STARTVerso3 is a Phase III trial that assessed the efficacy and safety of faldaprevir (FDV), a potent HCV NS3/4A inhibitor, plus pegylated interferon alfa-2a and ribavirin (PR) in patients with chronic HCV genotype-1 (GT1) infection who had virologic failure to prior PR treatment (29). In this double-blind, partially placebo-controlled trial, 3 cohorts were enrolled: cohort 1 (prior relapse), cohort 2 (prior partial response), and cohort 3 (prior null response). Cohorts 1&2 were each randomized 1:2:2 to receive 48 weeks(W) of PR plus: placebo, 12W FDV 240 mg QD (FDV12W) or 24W FDV 240 mg QD (FDV24W). In cohort 1, FDV-treated patients were eligible to stop all treatment at W24 on achievement of early treatment success (ETS: HCV RNA<25 IU/mL detected or undetected at W4, and undetected at W8). Cohort 3 was randomized 1:1 to receive FDV12W or FDV24W with PR for 48W. 677 patients were treated; 60% male, 77% Caucasian, 18% Asian,40% ≥F3 fibrosis, 53% GT1b, 16% IL28B (rs12979860) CC. FDV plus PR was most effective in HCV GT1 treatment-experienced patients with prior relapse reaching a SVR12 rate of 70%. Lower response rates were seen in prior partial responder with a SVR12 rate of 58% and in previous null responders with 33%. Treatment response rate dropped even further in cirrhotic previous null responders (between 13-18%). Treatment with FDV 240 mg for 12W was as effective as for 24W. Treatment discontinuation occurred in 7% of study subjects. Adverse event rates were higher in he 240mg faldaprevir arms of the study particularly with regard to hyperbilirubinemia. These results highlight that this particular combination will not be sufficient for treating more difficult patient populations such as cirrhotic or previous null responders to dual therapy.
The next pooled analyses presented was a retrospective analysis of data from 982 patients with HCV genotypes 1-6 treated with sofosbuvir (SOF) combination therapy in 4 Phase 3 clinical trials (FISSION, POSITRON, FUSION, and NEUTRINO) (30). The entry criteria of the Phase 3 studies SOF + ribavirin (RBV) with or without peginterferon were designed with minimal exclusions to safely enroll patients representative of the real-world HCV population which allowed for sub analyses in more challenging to treat populations including cirrhotics.. Overall, 21% of the patients had cirrhosis, 5% were ≥65 years, 33% obese (BMI ≥30), 14% had IL28B TT genotype, 8% had diabetes (HbA1c ≥6.5), 7% were black (with the highest percentage (17%) in the NEUTRINO study in genotype 1 patients), 16% had very high viral load at baseline (≥107 IU/mL), and 6% were on opiate replacement therapy. Figure 22 shows some of SVR 12 responses for the different patient subgroups tested.
Figure 22: SOF Phase 3 Analysis in Patients with Traditional Negative Factors
Virologic Response: SVR12 in NEUTRINO GT 1,4,5,6
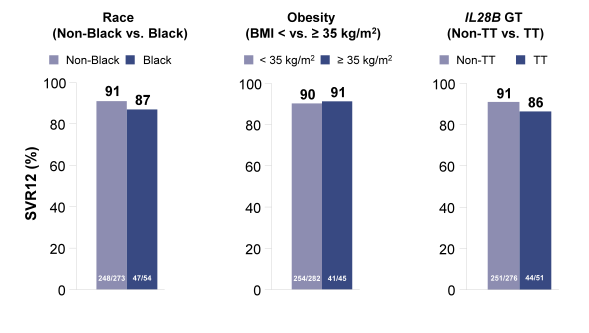
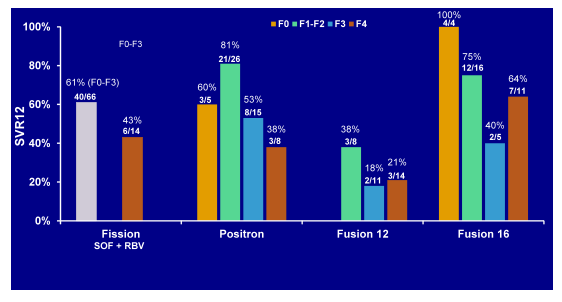
Reassuringly, race, obesity and IL28B GT had no impact on cure rates. Same was true for the IFN-free sofosbuvir+ribavirin treated GT2 patients. In conclusion, traditional negative predictive factors did not have a consistent influence on response rates with the exception of cirrhosis (lower SVR rates) and sex (females having higher cure rates than men). The impact of baseline fibrosis degree and extent of thrombocytopenia on response to therapy was further evaluated in an additional poster by Patel et al. again performing a retrospective analyses with the data from four Phase 3 clinical trials (FISSION,POSITRON, FUSION, and NEUTRINO) (31). Of 982 patients treated with a SOF-containing regimen, 857 (87%) had documentation of fibrosis stage by Fibrotest: 55% F0-F2, 17% F3, 28% F4. Thrombocytopenia (platelets <125/mm3) at baseline was present in 12% of patients, and 5% had platelets <100/mm3. SOF was well tolerated across all subgroups, including patients with cirrhosis and/or thrombocytopenia. Figure 23a shows the SVR12 rate depending on fibrosis stage for GT1,4,5, and 6 from the NEUTRINO trial and Figure 23b shows the SVR12 rate for GT3 patients from the FISSION, POSITRON and FUSION trial.
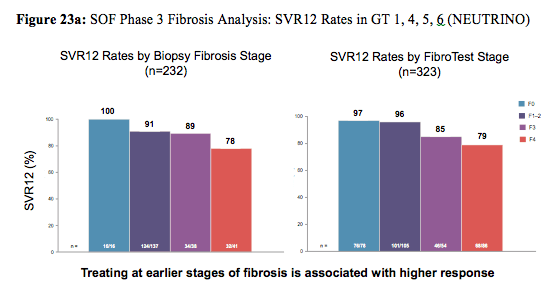
Figure 23b: SOF Phase 3 Fibrosis Analyses: SVR12 by Biopsy Stage in GT 3 Patients
In addition to the retrospective pooled data analyses with the upcoming DAAs there were also new DAA plus PEG-IFN/RBV studies which were presented at AASLD and which will be summarized below.
The most important study to be mentioned was the late breaker presentation of the LONESTAR-2 trial, which was an open-label, phase 2 study of the efficacy of SOF + pegylated interferon (PegIFN) + RBV for 12 weeks in treatment-experienced patients with GT 2 or 3 (32). As particularly patients with GT3 seem to benefit less from an all oral sofosbuvir + ribavirin combination, particularly in more advanced fibrosis stages or previous non-response to dual therapy, this study was helpful to understand how well more challenging GT3 might respond with interferon additionally on board. The study design is depicted in Figure 24.
Figure 24: Study design of the LONESTAR-2 trial
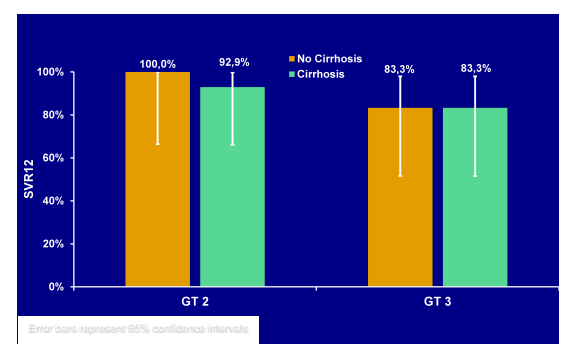
Treatment-experienced GT2/3 HCV-infected patients, the majority (55%) of whom had cirrhosis, were enrolled in a single arm, open-label study and received SOF 400mg daily + PegIFN 180μg weekly +RBV 1000-1200mg daily for 12 weeks. Overall, 47 patients with GT (n=23) or GT3 (n=24) were enrolled. An impressive 55% had compensated cirrhosis and 36% were genotyped as IL28BCC. The obtained SVR12 rates by genotype and baseline presence of cirrhosis are shown in Figure 25.
Figure 25: SVR12 rates from the LONESTAR-2 trial for GT 2 and 3 patients with and without cirrhosis.
The adverse event (AE) profile was consistent with that observed with PegINF+RBV. The most common AEs were: flu-like symptoms (55%), fatigue (32%), anemia (30%), neutropenia (23%), and nausea (17%). SAEs occurred in 4 (9%) patients with no individual SAE occurring in >1 patient. Overall, the high response rates irrespective of baseline cirrhosis suggest that indeed the addition of PEG-IFN to sofosbuvir and RBV can substantially consolidate response rates in these more challenging to treat patients.
Interfeon-free therapies
1 DAA + Ribavirin
The VALENCE trial was a phase 3, randomized, safety and efficacy study of all-oral sofosbuvir (SOF) + ribavirin (RBV) for 12 or 24 weeks in treatment-naïve (TN) or -experienced (TE) patients infected with HCV genotype (GT) 2 or 3 which was largely done in Europe as most Sofosbuvir studies so far had been done exclusively in North America or New Zealand (33). Treatment-naïve or treatment-experienced patients infected with HCV genotype 2 or 3 were randomized 4:1 to receive SOF+RBV for 12 weeks or matching placebo. The study was subsequently amended to extend treatment duration to 24 weeks for patients with genotype 3 HCV irrespective of prior treatment history. Therefore, this was the first study to address the question whether extending treatment duration to 24 weeks in GT 3 patients would prevent relapses and increase overall SVR rates. The study design of the VALENCE study is shown in Figure 26.
Figure 26: Study design of the VALENCE study
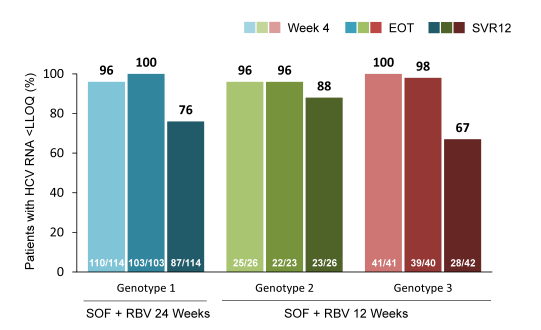
Overall, 421 patients were randomized: 334 received SOF+RBV, 85 received placebo, and 2 did not initiate treatment. Of those randomized to receive SOF+RBV, 261 (78%) had HCV genotype 3, 195 (58%) were treatment-experienced, 70 (21%) were cirrhotic, and 114 (34%) had the IL28b CC genotype. All patients receiving SOF+RBV became and remained HCV RNA negative while on treatment; relapse has accounted for all virologic failures to date. The SVR12 rates for the different genotypes (for TN and TE patients as well as depending on baseline cirrhosis) are depicted in Figure 27a and b. The therapy of SOF+RBV for 12 or 24 weeks was generally well tolerated and only 2 (<1%) patients have discontinued treatment early due to adverse events (malaise and headache in one patient, and suicide attempt in the other). Adverse events and laboratory abnormalities were generally consistent with the safety profile of RBV. The most frequent adverse events in patients who received SOF+RBV were: headache, 28%; fatigue, 27%; pruritus, 24%; asthenia, 22%; nausea, 17%; insomnia, 14%; dyspnea, 11%; and dry skin, 11%. Extending treatment duration to 24 weeks did not increase the incidence of AE.
Figure 27a: SOF + RBV for 12 Weeks for HCV GT 2 (VALENCE trial) Virologic Response and SVR12
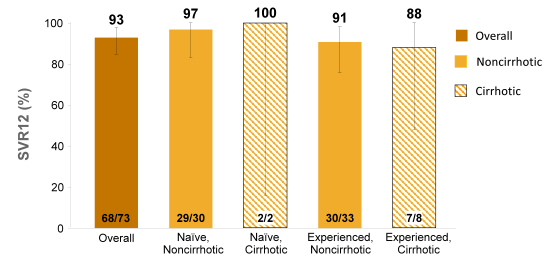
Figure 27b: SOF + RBV for 24 Weeks for HCV GT 3 (VALENCE trial) Virologic Response and SVR12
In summary, efficacy in genotype 2 patients was similar to that observed in recent phase 3 studies. Data on the efficacy of SOF+RBV for 24 weeks in genotype 3 HCV-infected patients is critical to optimize the treatment duration for HCV genotype 3 infections and offer improved SVR rates. More challenging GT 3 patients (such as treatment experienced and cirrhotic) may require DAA combination therapy in the future.
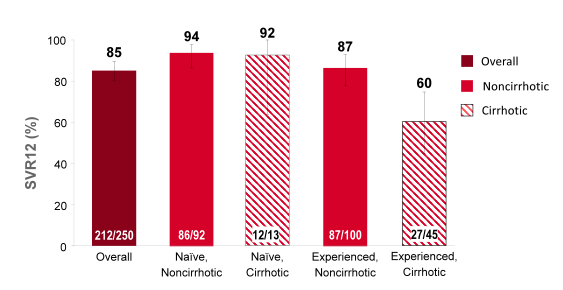
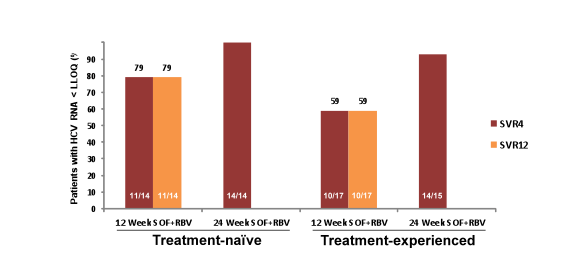
Another sofosbuvir study evaluated efficacy and safety of the sofosbuvir + ribavirin combination this time in GT4 patients (34). So far data for treatment of GT 4 is rare but in Europe GT4 infections are increasing due to migration and clearly improved treatment options are needed here as well. In this pilot trial from the US treatment-naive and treatment-experienced patients with HCV GT4 infection were randomized 1:1 to receive either 12 or 24 weeks of SOF (400 mg daily) + RBV (1000-1200 mg daily). Patients were born in Egypt, with both maternal and paternal Egyptian ancestry documented. Randomization was stratified by prior treatment experience and by the presence or absence of cirrhosis. The study design is shown below in Figure 28.
Figure 28: Randomized, open-label, single-center study conducted in the US of the safety and efficacy of all-oral SOF + RBV in patients of Egyptian ancestry with HCV GT 4
60 patients were enrolled (28 treatment-naive and 32 treatment-experienced). Among treatment-experienced patients, 62.5% were prior non-responders to therapy, 18.8% had experienced relapse or breakthrough on treatment, 12.5% were interferon-intolerant, and prior response was unknown for 6.2%The study population in the two study arms had a mean age of 52-57 years, mean BMI was 28-31 kg/m2, 21-27% had cirrhosis at baseline and 57-100% were L28B non-CC. The interim virological response rates are shown in Figure 29. Overall high SVR4 rates were observed for TN as well as TE patients. However, it looks as if longer duration of combination treatment with sofosbuvir and ribavirin was associated with better SVR rates in TN and even more pronounced in TE patients with GT4 infection.
Figure 29: Virological response
The C-SPIRIT trial investigated the interferon-free, single DAA regimen of MK-5172 (a potent, pan-genotypic, second generation hepatitis C virus (HCV) NS3/4A protease inhibitor with a high genetic barrier to resistance) +ribavirin (RBV) in patients with HCV GT 1 infection who express the IL28B CC genotype (35). Obviously this is a relative easy to treat patient population because of the favorable CC IL28b genotype distribution but clearly will help us to answer the question whether easier to treat patients could be treated with one potent DAA + ribavirin sufficiently which could have a great cost impact.
Treatment-naive, non-cirrhotic, IL28B CC genotype patients with HCV GT1 infection were randomized to receive 12 or 24 weeks of MK-5172 100 mg QD + weight-based RBV BID. Patients in the 12-week arm with detectable HCV RNA at treatment week (TW) 4 had their treatment duration extended to 24 weeks. 12/25 patients had the GT1a subtype. The most frequently reported adverse events (>10%) were headache (6, 23%), asthenia (6, 23%), anemia (3, 12 %), dyspepsia (3, 12%), nausea (3, 12%), and insomnia (3, 12%). There were no SAEs or treatment discontinuations. ALT was elevated at baseline in 16 patients (range, 34-252), but normalized on treatment in all patients. Nine patients had transient, mild (grade 1-2) elevations in total bilirubin on treatment (range, 1.12-2.45). Good early response rates were observed in all patients with 100% of patients being undetectable at week 4 of therapy. At EOT 83% of the GT1a and 92% of the GT1b patients remained undetectable, respectively. SVR4 so far was 73% for GT1a and remained 92% for GT1b. These results show very good efficacy for this HCV-PI + RBV combination for GT1b. GT1a patients respond less favorably although SVR rates are still high.
2-3 DAA +/- Ribavirin
One of the most interesting studies looking at 2 DAAs +/- ribavirin was the COSMOS study (36). This study is of particular interest as it evaluated the efficacy and safety of the two DAAs sofosbuvir and simeprevir plus/minus ribavirin which will be the next DAAs which will become available in North America, Japan and Europe. Obviously this prompts the question whether this may be a good combination for more difficult to treat patient populations who are unable to take an interferon-containing HCV regimen. Now as the two drugs are being developed from 2 different companies there is no planned phase III program but this study was already started early in the sofosbuvir development history when the drug had not yet been sold to a new company. (36). The COSMOS study was a Phase II, randomized, open-label study investigating efficacy/safety of SMV+SOF±ribavirin (RBV) for 12/24 weeks (weeks) in HCV genotype 1(GT1) patients (pts) with METAVIR score F0-F2 who were prior null responders to PegIFN/RBV (cohort 1, C1) or treatment-naïve patients and prior null responders with F3-F4 (cohort 2, C2). The cohorts were enrolled sequentially. Patients were randomized to SMV (150mg QD)+SOF (400mg QD)±RBV for 12 weeks, or SMV+SOF±RBV for 24weeks. C1 was stratified by GT1 subtype and IL28B, and C2 by GT1 subtype and prior treatment status. The virological outcome for cohort 1 ad 2 is shown in Figure 30a and b.
Figure 30a: Virological response (SVR12) in cohort1 (Null responders with METAVIR F0-2)
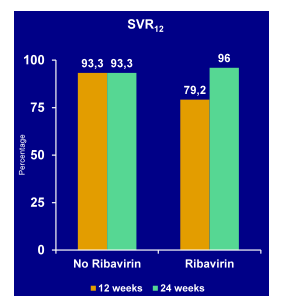
Cohort 1 consisted of prior Null responders with METAVIR F0-2. Patients were mostly Caucasian (only 28.8% were Black), 77% had a genotype 1a, 50%; had a Q80K at baseline, 6% were IL28B CC, and 59% had a F2-fibrosis. Cure rates were high with or without ribavirin and independent from 12 or 24 weeks of treatment duration.
Figure 30b:Virological response (SVR4) in cohort 2 (treatment-naïve patients and Null responders with METAVIR F3-4)
Cohort 2 (n=84) consisted of treatment-naïve and null responders with METAVIR F3-4 the majority (78%) of patients had genotype 1a, 40% had a Q80K at baseline, 47% were cirrhotic and 54% had a previous null response to dual therapy. Even in this more difficult to treat patient population including a fair amount of cirrhotic null-responders overall SVR4 rates were very high suggesting great efficacy of this combination. Also it appeared as if the addition of ribavirin did not add much in terms of cure rates, again highlighting that DAA combinations may emerge were ribavirin can be left out which would be highly attractive with regard to its safety profile plus inability to use it in renal insufficiency. As SVR12 rate are still pending more late relapses cannot be precluded so waiting for the final results for final interpretation is recommended.
AASLD: SVR results of a once-daily regimen of simeprevir (SMV, TMC435) plus sofosbuvir (SOF, GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naïve and prior null responder patients: The COSMOS study - (11/05/13)
In the SOUND-C2 study the efficacy and safety of faldaprevir, twice-daily deleobuvir (BI 207127; a nonnucleoside polymerase inhibitor) and ribavirin were evaluated. Within this study an induction dose of deleobuvir was given on Day 1 leading to high plasma concentrations and increases in gastrointestinal adverse events (AEs). At AASLD the SOUND-C3 was presented which investigated the same regimen, but without an induction dose of deleobuvir, dosed for 16 weeks in GT1b or GT1a-CC patients (37). Patients infected with GT1b (n=20) or GT1a-CC (n=12) received faldaprevir 120 mg once-daily, deleobuvir 600 mg twice-daily and weight-based ribavirin for 16 weeks. Patients with compensated cirrhosis were included in the study (n=4 in the GT1b arm). The virological response rates of this pilot trial are shown in Figure 31.
Figure 31: Virological response in SOUND-C3: Faldaprevir + Deleobuvir + Ribavirin for Patients with Genotype 1a/IL28 CC and 1b
Viral breakthrough (n=4) and relapse (n=6) occurred mainly in GT1a patients (9 of 10 had all GT 1a). Tolerabilty was good with only 3 discontinuations due to AE (1 due to nausea/vomiting, 1 because of increase in bilirubin). All 20 patients with GT1b and 8/12 of patients with GT1a-CC had undetectable HCV RNA at the end of treatment. SVR12 was achieved in 19/20 patients with GT1b. The only GT1b patient without SVR12 was a non-cirrhotic patient who discontinued treatment at Week 8 with undetectable HCV RNA and relapsed 12 weeks later. In contrast, only 2/12 of patients with GT1a-CC achieved SVR12, with 6/8 patients with end-of-treatment response experiencing a relapse. Overall tolerability was good with 4/32 severe treatment-related AEs, 1/32 serious AE (dehydration requiring hospitalization) and 3/32 early discontinuations due to an AE (dehydration, vomiting and pruritus). In a patient with virological failure the D168A/N mutation was observed. Overall, it appears that this combination might work very well in GT1b but may not be sufficient for GT1a patients. Therefore this combination will be tested in phase III in GT1b patients only.
Boehringer also reported back from a Phase 2 trial of a new 3-class oral regimen, combining PPI-668 (NS5Ainhibitor) with faldaprevir and deleobuvir (a nonnucleoside polymerase inhibitor) in treatment-naïve patients with HCV GT-1a infection (38). Overall 36 patients were included into the study. 24 were randomized (1:1) to Cohort 1 (faldaprevir 120 mg QD, PPI-668 200mg QD, and deleobuvir 600 mg BD, plus weight-based RBV) or Cohort 2 (same doses of faldaprevir, PPI-668, and RBV, with a lower 400 mg BD dose of deleobuvir). A third 12-patient cohort was enrolled to study a RBV-free regimen that is otherwise identical to Cohort 1. Key entry criteria were HCV RNA >5 log10 IU/mL (HCV TaqMan™ 2.0, assay), HCV GT-1a, and no known cirrhosis. Enrollment of patients with the 'favorable' IL28B genotype CC was limited to one third of the population, to ensure a more difficult-to-treat population. The HCV RNA reductions for the 3 different cohorts are shown in Figure 32.
Figure 32: Mean HCVlog10 RNA reduction from baseline
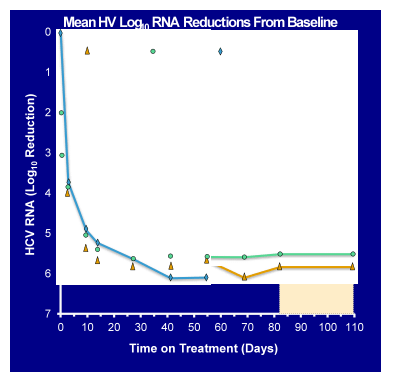
Overall, in this GT1a population 97% of patients across all three cohorts achieved HCV-RNA < LLOQ (81% < LLOD) at week 4 regardless of ribavirin use. These data strongly suggest that with the addition of the NS5A-inhibitor PPI-668 this 3DAA combination in contrast to the SOUND-C2 study (only 2DAAs: faldaprevir and deleobuvir + ribavirin) exerts strong antiviral potency even in GT1a patients. Most interestingly, even patients with baseline detectable resistance substitutions or polymorphisms responded well in the study to date. Clinical adverse events have been similar to those previously seen in studies with faldaprevir and deleobuvir (mainly skin rashes and GI side effects). Patients from the ribavirin-free arm seemed to have fewer side effects than patients from the ribavirin-containing arms. Overall, only one treatment discontinuation due to an AE occurred (due to GI side effects). In conclusion, this new 3 DAA combination, even without ribavirin, produced a very promising rapid virological response. The first patients reaching the SVR4 time point have remained undetectable. Clearly, longer follow-up is needed to have data from all patients available to be able to interpret the final SVR results.
The C-WORTHY study aimed to assess the efficacy and safety of a 12-week regimen of MK-5172 (a potent hepatitis C virus [HCV] NS3/4A protease inhibitor) and MK-8742 (a potent HCV NS5A replication complex inhibitor) ± ribavirin (RBV) in patients with genotype (GT) 1 HCV infection (39). In in vitro studies, both agents have demonstrated a high barrier to resistance and activity against GT1 variants that are resistant to other first-generation agents in the same classes. Therefore MK-5172 has also been considered a true second generation HCV protease inhibitor as it may work even in the presence of PI resistance from first generation HCV PIs. Treatment-naive, non-cirrhotic (F0-F2) patients with HCV GT1 infection were randomized to one of three 12-week treatment arms: 1. MK-5172 (100 mg QD), MK-8742 (20 mg QD) plus RBV (600-1400 mg/d); 2. MK-5172 (100 mg QD), MK-8742 (50 mg QD) plus RBV; 3. MK-5172 (100 mg QD) and MK-8742 (50 mg QD). Arms 1 and 2 were stratified by GT1a vs. GT1b. Arm 3 (RBV-free) included only GT1b-infected patients. A total of 65 patients were enrolled: 45% were male, 11% were African American, and 58% had GT1a infection. Mean baseline HCV RNA was 6.1 log10 IU/mL. The most frequently reported adverse events (>10%) were headache (13/65, 20%), fatigue (15/65, 23%), and nausea (8/65, 12%). ALT was elevated at baseline in 50 patients and normalized on treatment in all but 1 patient (ALT = 42 IU/mL). Twenty patients had transient, mild (grade1-2) total bilirubin elevation on treatment. No discontinuation because of adverse events was noted during the trial. The virological response rates are shown below in Figure 33.
Figure 33: Virological responses (EOT/SVR12) in the C-WORTHY Study: MK-5172 + MK-8742 with or without RBV in genotype 1
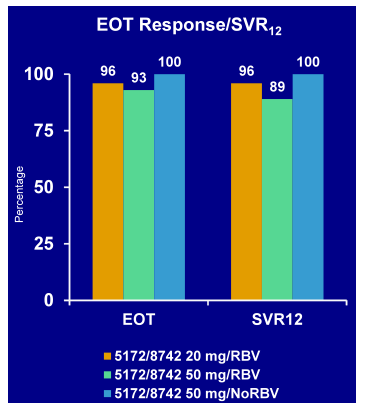
Clearly overall, impressive SVR rates were obtained in this first all oral combination of MK-5172 + MK-8742 in GT1 patients. Additional ribavirin is not needed for treatment of GT1b patients. It will be very interesting to see how this combination may perform in a more challenging patient population of cirrhotics and null-responders.
In the ELECTRON Phase 2 study the safety and efficacy of a fixed-dose combination (FDC) tablet of sofosbuvir (SOF) plus ledipasvir (LDV) (SOF 400 mg/LDV 90 mg one co-formulated tablet QD) was evaluated in additional new patient populations with cirrhosis and prior non-response to dual HCV therapy with GT-1 infection (40). The study also investigated the need for RBV or a third DAA GS-9669 (a QD non-nucleoside polymerase inhibitor and the optimal treatment duration. Overall, 5 arms were enrolled: prior non-responder HCV GT-1 patients with compensated cirrhosis were randomized to receive open-label SOF/LDV FDC with or without RBV for 12 weeks; prior non-responder HCV GT-1 patients with F3/F4 fibrosis to SOF/LDV FDC with ribavirin or GS-9669 and treatment-naïve HCV GT-1 patients without cirrhosis received SOF/LDV FDC plus RBV for 6 weeks. The study design is depicted below in Figure 34
Figure 34: Study design of the ELECTRON study
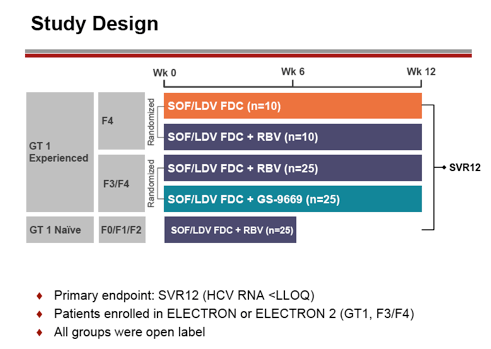
95 patients were enrolled. Of the GT-1 treatment experienced group, 60-85% were genotype 1a, and 32% were IL28B CC. Of the treatment-naïve GT-1 group, 84% were genotype 1a, and 15-40% were IL28B CC. Of the treatment-naïve GT-1 group 84% were GT1a and 20% IL28B CC. The virological response rates for the different groups respectively are shown in Figure 35.
Figure 35: SVR12 results: GT1 treatment experienced patients with advanced fibrosis/cirrhosis.
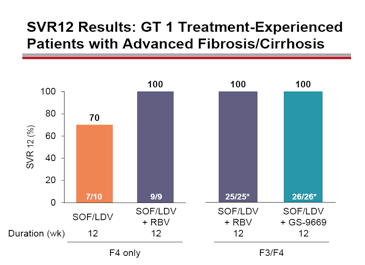
The response rates indicate that in F4 fibrosis the addition of ribavirin to the FDC of SOF/LDV enhances cure rates substantially. Alternatively rather than administering ribavirin the addition of a 3rd DAA (here GS-9669) also helps to achieve 100% SVR rates. The treatment arm with the treatment-naïve GT1 patient population GT 1(n=25, F≤2) which received SOF/LDV + RBV for 6 weeks revealed a SVR12 rate of 68% (17 of 25). 2 of 8 patients which presented with relapse had NS5A RAVs. This clearly indicates that 6 week treatment duration is not long enough for a substantial proportion of patients. Longer treatments durations of 8 and 12 weeks respectively had led to 100% SVR12 rates previously. Overall, the therapy was well tolerated with no treatment discontinuations due to adverse events.
Another study evaluating the safety and efficacy of 8 and 12-week regimens of the fixed-dose combination (FDC) of SOF and LDV with and without RBV in treatment-naïve and protease inhibitor-experienced patients with HCV genotype 1 was the LONESTAR trial (41, 42). The study design is shown below in Figure 36:
Figure 36: Study design of the LONESTAR trial
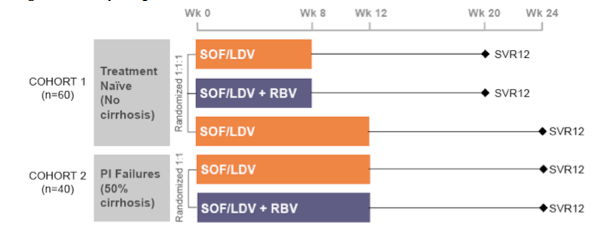
60 non-cirrhotic treatment-naïve patients with HCV genotype 1 were randomized 1:1:1 to receive: 1) FDC for 8 weeks, 2) FDC + RBV for 8 weeks, or 3) FDC for 12 weeks. In parallel, 40 patients who had not achieved SVR after previous treatment with a protease inhibitor regimen (50% of whom also had compensated cirrhosis) were randomized to receive twelve weeks of: 1) FDC or 2) FDC + RBV. Within the total patient population of 100 87% had a GT1a and 15% were IL28B CC. Of the PI pretreated patients 55% had been treated with boceprevir, 45% with telaprevir. The virological outcome is summarized in Figure 37. Again tolerability was very good with no treatment discontinuations because of adverse events.
Figure 37: SVR12 results for the different cohorts from LONESTAR
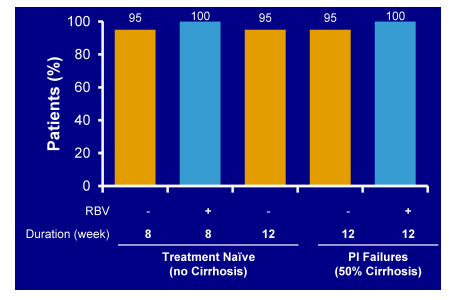
An impressive 97/100 achieved SVR12 regardless of baseline cirrhosis or previous failure to HCV PI based triple therapy. Of the 3 patients non-responding one patient was lost to follow-up, the other two developed a relapse. Potentially 8 weeks of therapy could be sufficient in treatment-naïve patient populations. For non-responders to HCV PI based triple therapy no shorter durations than 12 weeks have been investigated so far. Most interestingly, there was one patient who relapsed in the 8 week SOF/LDV arm who had developed NS5A and NS5B relevant mutations but was successfully retreated with SOF/LDV/RBV. This implies that even patients who do not respond to a simpler ribavirin free regimen can potentially subsequently be rescued with the addition of ribavirin.
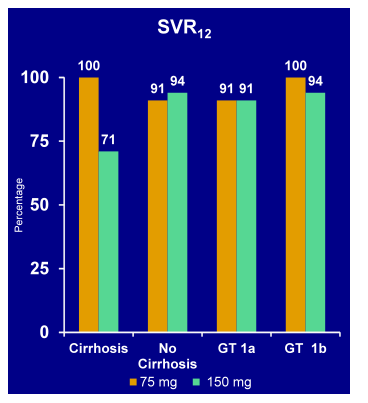
Finally, a new analysis from the AVIATOR study was presented (43). In this new sub analyses the safety of a RBV-containing, 3-DAA (ABT-450/r (HCV protease inhibitor dosed with ritonavir 100 mg), ABT-267 (NS5A inhibitor), and ABT-333 (non-nucleosideNS5B inhibitor), pegIFN-free regimen and the impact of RBV dose reduction on treatment response was investigated. This study looked at a subset of treatment naïve (n=156) and null-responders (n=88) treated for 12 or 24 week with the three DAAs/RBV. Four patients (1.6%) discontinued due to study drug -related AEs, and 1 patient (0.4%) had a serious AE (arthralgia) considered possibly related to study drug. Hemoglobin values <10 g/dL and <8.5 g/dL occurred in 16 (6.5%) and 1 (0.4%) subjects during treatment, respectively. RBV dose was reduced in 27 subjects (10.9%) due to toxicity. Sixteen of the 27 reductions were due to anemia AEs. Other AEs, including diarrhea, fatigue, increased blood creatinine, and dizziness, led to RBV dose reductions less frequently. The overall SVR rates for the patients with and without ribavirin dose reductions are shown in Figure 38.
Figure 38: AVIATOR Study: Ribavirin Dose Reduction in GT 1 Treated with ABT-450/r, ABT-333, ABT-267 + RBV: SVR24 rates
RBV dose reductions were required less frequently with this pegIFN-free regimen than in previously reported studies of subjects receiving pegIFN-containing regimens. High SVR12 rates (100%) were achieved among subjects requiring RBV dose reduction.
2 DAA without Ribavirin
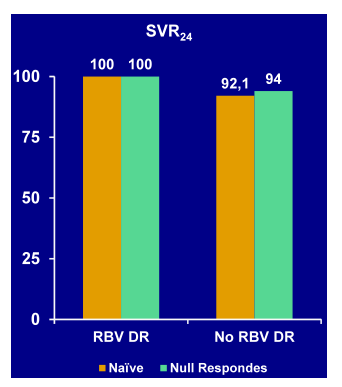
The first important study to report on is an open-label, phase III trial evaluating the safety and efficacy of a PEG/RBV-free, dual oral therapy with daclatasvir (DCV, NS5A inhibitor) and asunaprevir (ASV; HCV PI) in interferon ineligible naive/intolerant (IN/I) and non-responder (NR) Japanese patients infected with HCV genotype 1b (44). As in Japan the HCV GT1b is by far the most common genotype found and as 1b is in general easier to treat, different treatment regimens may work in this particular population as in a more typical North American patient population with mostly GT1a and a less favorable IL28b genotype distribution than in Japan. In this open-label, parallel group, phase 3 study, IN/I (n=135) and NR (n=87) patients received DCV 60 mg once daily plus ASV 100 mg twice daily for 24 weeks. The median age of patients was 62.5 years, 65% were female, and 10% were cirrhotic Mean baseline HCV RNA was 6.6 log10IU/mL. IN/I patients were primarily CC IL28B genotype (70%), 82% of NR patients were non-CC IL28B genotype (rs12979860). The virological response rates for the different treatment populations are shown n Figure 39:
Figure 39: Virological response: EOT and SVR 24 from a phase III trial with daclatasvir and asunaprevir
The most common AEs were nasopharyngitis (30%) increased ALT (16%) and AST (13%),
headache (15%), diarrhea (10%), and pyrexia (10%). Most discontinuations were due to ALT/AST elevations. Clearly hepatoxicity potential of this combination warrants close monitoring and larger studies will be needed to receive a better idea of the magnitude and clinical implications of these liver enzyme elevations. Response rates are high although numerically there is a decline in SVR rate in previous non-responders. Obviously interferon and ribavirin free regimens are highly desirable to get rid of the safety issues associated with the two compounds.
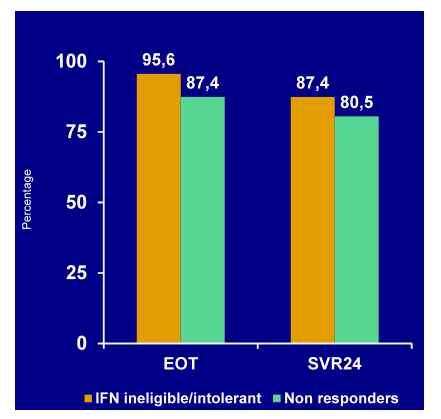
The PEARL-I study is evaluating the safety and efficacy of an IFN-and RBV-free regimen of ABT-450/r (an HCV protease inhibitor dosed with ritonavir 100mg), +ABT-267 (an NS5A-inhibitor in patients with HCV GT1b infection (45). Non-cirrhotic, treatment-naïve patients and prior pegIFN/RBV null responders received ABT-450/r(150/100mg QD) and ABT-267(25mg QD) for 12 wks. This is also interesting with regard to the fact that most Abbvie phase III studies additionally have ABT-333(a non-nucleoside polymerase inhibitor) on board and indeed the question may arise whether a combination of 3 DAAs is really needed for all HCV patients. Therefore this study allows to assess the efficacy of a 2DAA regimen in a more easy to treat patient population (no cirrhosis, GT1b). It is important to highlight though, that non-responders were also included into this trial which obviously are more challenging to cure. Overall, 42 treatment-naïve patients and 40 prior null responders with chronic HCV GT1b infection were enrolled. The virological response rates are shown in Figure 40
Figure 40: Virological response rates (EOT/SVR12)
The high SVR rates in treatment naïve, non-cirrhotic GT1b patients indeed suggests that simpler combinations will become applicable for easier to treat patient populations. Even for non-responders results were quite impressive although somewhat numerically lower suggesting that there could be a subgroup requiring 3 DAAs or addition of ribavirin. Safety data was also promising with only 2 subjects interrupted study drug due to AEs. 1 interruption was considered probably related to study drug (increased ALT, AST, and bilirubin) which warrants further follow-up.
3 DAA without Ribavirin
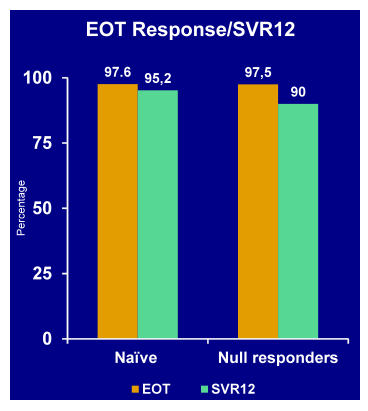
The all-oral triple combination of daclatasvir (DCV; NS5A inhibitor), asunaprevir (ASV; NS3 inhibitor), and BMS-791325 ('325; non-nucleoside NS5B inhibitor) has been demonstrated to achieve sustained virologic response (SVR) rates >90% in pilot cohorts of non-cirrhotic patients with HCV genotype (GT) 1 infection. At this year AASLD the study expansion (AI443-014) which evaluated this 3 DAA regimen in larger cohorts that included cirrhotic patients was presented (46). 166 treatment-naïve, HCV GT1-infected patients were randomly assigned (1:1) to receive a twice-daily regimen of DCV 30mg, ASV 200mg, and "325" 75mg (n=80) or 150mg (n=86) for 12 weeks. Randomization was stratified by GT1 subtype and presence of cirrhosis. The virological outcome results are summarized in Figure 41.
Figure 41: SVR12 rates in HCV GT1 patients Daclatasvir + Asunaprevir + BMS-791325
for 12 Weeks in Treatment-naïve with Genotype 1
The 12 week, IFN and ribavirin free all oral 3 DAA regimen with DCV/ASV/BMS-791325 achieved SVR12 in >90% of patients despite prevalence of GT1a or advanced fibrosis/cirrhosis. The therapy was overall well tolerated with low rates of adverse events and treatment discontinuations, regardless of BMS-791225 dose. One patient however developed a grade 3/4 AST elevation which needs to be followed carefully. These findings support the initiation of phase 3 trials with a twice daily fix-dose combination of DCV/ASV/BMS-791325 at the 75mg dose level; in this phase 3 study a ribavirin add-on is included.
Finally in the NIAID SYNERGY Trial 60 HCV mono-infected, treatment naïve, GT-1, patients were consecutively enrolled into 3 arms of a phase 2 clinical trial and received: Arm A - sofosbuvir with ledipasvir (400mg/90mg respectively once daily in a fixed dose combination (FDC)) for 12 weeks, Arm B - FDC + GS-9669 (500mg/day), a non nucleoside NS5B inhibitor for 6 weeks, or Arm C - FDC + GS-9451 (80mg/day), an HCV protease inhibitor for 6 weeks (47). 80-95% of study participants were African-American. In this exploratory trial virological responses were very good with 100% SVR12 in the SOF/LDV (12 weeks arm) (n=20), 90% SVR4 in the SOF/LDV/9669 (6 weeks arm) (n=20) and again 100% SVR4 in the SOF/LDV/9451 (6 weeks arm) (n=20). Only one relapse occurred in the SOF/LDV/9669 arm. Tolerability in all arms was good with no treatment related discontinuation or SAEs. Again this 3 DAA combination study underlines that multiple DAA combinations will work and allow to get rid of interferon as well as ribavirin which is accompanied by greatly improved tolerability. Also the combination of 3 DAAs may allow to shorten treatment duration to 6 weeks at least in easier to treat naïve patient populations which may be very important in the more adherence challenged patient groups.
Summary
· During 2005-2011 at 8 US surveillance sites 217,755 persons were identified as newly HCV positive, whereby 49% lacked HCV-RNA testing. These findings underline that within the HCV treatment cascade not only testing but also linkage to care following new HCV diagnosis in the US remains a huge challenge.
· The safety profile of TVR or BOC with PEG-IFN/RBV in real-life cohorts evaluating cirrhotic patients is poor and is associated with increased discontinuation rates due to SAEs compared to those reported in phase III trials. In particular, patients with platelets below 110.000/μl and lower albumin levels < 35-40 g/l are at high risk of SAEs and should only be treated in very experienced centers. Cure rates are lower in the context of cirrhotic non-responders but are higher than 50% in previous relapsers. These data strongly suggest that triple therapy must be administered cautiously with intensive safety monitoring in patients with liver cirrhosis.
· Studies in HIV/HCV coinfected previous non-responders to dual therapy with PEG-IFN/RBV and a considerable proportion of patients with more advanced fibrosis stages demonstrate very high on treatment response rates under telaprevir or boceprevir based triple therapy. Overall, it appears as if boceprevir works less well in the context of previous non-responders or breakthroughs. These results should remind us not to postpone HCV therapy for all in hope of easier and less toxic HCV treatment regimens in the future but also to at least consider the improved treatment options we already have in our hands which preferably should be used in those patients where risk for development of HCC or progression to cirrhosis and eventually hepatic decompensation is a real threat.
· Faldaprevir plus PegIFN/RBV in HIV/HCV coinfected HCV treatment-naïve or relapser patients promises good efficacy for most patients with short treatment durations following response guided therapy. This is the first HCV PI which can be administered concomitantly with boosted darunavir.
· Results from the first interferon-free trial in HIV/HCV coinfection demonstrate that the majority of GT1may be curable with the all oral sofosbuvir plus ribavirin combination for 24 weeks. Clearly 12 weeks of this combination are sufficient for treatment in GT 2 patients. For GT 3 patients 12 weeks appear to be to short and longer treatment durations need to be evaluated in this patient population. There will be a need for DAA combination therapy in around a quarter of GT 1 patients reflecting the relatively high relapse rate after treatment discontinuation.
· A triple therapy regimen including a PI for recurrent genotype 1 HCV in liver transplant recipients appears significantly more effective than treatment with P/R only, with projected SVR rates between 48 and 58%. Adverse events are however common, requiring careful monitoring and mostly intensive anemia management including EPO, transfusions and ribavirin dose reductions.
· Pre-transplant therapy with Sofosbuvir and RBV are safe and effective in patients with well compensated cirrhosis, and prevented post-transplant HCV recurrence in 69% of patients who had HCV RNA <25 IU/mL prior to transplant. Main predictor for recurrence was time being undetectable for HCV.
· Administration of SOF and RBV after liver transplantation in the setting of established HCV recurrence has been well tolerated and achieved an early SVR4 rate to date of 77%. There were no episodes of rejection or drug interaction.
· Pooled analyses from Simeprevir phase III trials conferred clinical benefit across all patient sub-populations, including not only those with positive predictors of response (IL28B CC, METAVIR F0-F2, GT 1band GT 1a/Q80K-), but also patients with unfavorable baseline characteristics, such as IL28B TT, METAVIR F4 and GT 1a/Q80K+. Discontinuation rate because of adverse events was low with 3% in the pooled analyses.
· Overall, simeprevir/PEG-IFN/RBV treatment promises easier HCV therapy than with telaprevir or boceprevir as simeprevir is dosed once daily. The safety also looks better than previous triple therapy. Concerns remain with regard to efficacy in patients with a baseline Q80K which was shown to be present in 13.7% of all HCV genotypes, but being found most frequently in genotype 1a patients.
· Overall, faldaprevir based triple therapy in HCV treatment-naive patients also allows for simplified therapy (faldaprevir is administered once daily) in comparison to telaprevir/boceprevir based HCV therapy with comparable high SVR rates.
· Pooled analyses from the sofosbuvir phase 3 trials suggested, that traditional negative predictive factors did not have a consistent influence on sustained virological response rates with the exception of cirrhosis (lower SVR rates) and sex (females having higher cure rates than men).
· SOF+PegIFN+RBV combination therapy for 12 weeks demonstrated high efficacy in treatment-experienced GT2/3 patients who have historically low response rates and limited treatment options.
· In the VALENCE trial efficacy in genotype 2 patients was similar to that observed in recent Phase 3 studies. Data on the efficacy of SOF+RBV for 24 weeks in genotype 3 HCV-infected patients is critical to optimize the treatment duration for HCV genotype 3 infections and offer improved SVR rates. More challenging GT 3 patients (such as treatment experienced and cirrhotic) may even require DAA combination therapy in the future to further improve SVR rates.
· SOF+RBV therapy for HCV GT4 leads to high SVR4 rates in treatment-naïve as well as treatment-experienced patients. However, it looks as if longer duration of combination treatment with sofosbuvir and ribavirin (for 24 weeks) was associated with better SVR rates in TN and even more pronounced in TE patients with GT4 infection.
· Obviously interferon and ribavirin free regimens are highly desirable to get rid of the safety issues associated with the two compounds. Studies looking at the combination of asunaprevir and daclatasvir or the combination of ABT-450/r + ABT-267 show good efficacy in GT1b patients.
· Impressive SVR rates were obtained in the first all oral combination of MK-5172 + MK-8742 in GT1 patients. Additional ribavirin is not needed for treatment of GT1b patients. It will be very interesting to see how this combination may perform in a more challenging patient population of cirrhotics and null-responders.
· In an update from the ELECTRON studies it was demonstrated that in F4 fibrosis the addition of ribavirin to the FDC of SOF/LDV enhances cure rates substantially. Alternatively rather than administering ribavirin the addition of a 3rd DAA (here GS-9669) also enhances cure rates. 6 weeks of SOF/LDV are associated with increased relapse rates compared to 8 or 12 weeks suggesting that this duration may be to short for a considerable proportion of treatment-naïve non-cirrhotic patients.
· The 12 week, IFN and ribavirin free all oral 3 DAA regimen with DCV/ASV/BMS-791325 achieved SVR12 in >90% of patients despite prevalence of GT1a or advanced fibrosis/cirrhosis. The therapy was overall well tolerated with low rates of adverse events and treatment discontinuations, regardless of BMS-791225 dose. These findings support the initiation of phase 3 trials with a twice daily fix-dose combination of DCV/ASV/BMS-791325 at the 75mg dose level.
· Addition of a third DAA to a FDC can lead to more rapid suppression of HCV VL and may achieve high rates of SVR with only 6 weeks of therapy, shortening the treatment duration for HCV and further simplifying management of HCV-infected subject
In summary, evidence is accumulating that with an increasing number of DAAs and combinations it appears very likely that there will be an interferon-free, most likely even a ribavirin free HCV combination for every challenging HCV patient group in the near future.
References
1. Sulkowski M: Hepatitis Debrief. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; Session: Hepatitis Debrief
2. Valdiserri R: Introduction to Hepatitis Debrief. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; Session: Hepatitis Debrief
3. Holtzmann D: et al. Vital Signs: Evaluation of Hepatitis C Virus Infection Testing and Reporting - Eight U.S. Sites, 2005-2011. MMWR 2013;62 (18):357-361
4. Tohme RA et al., Hepatitis C Testing, Infection, and Linkage to Care Among Racial and Ethnic Minorities in the United States, 2009-2010.,Am J Public Health 2013;103:112-119
5. Holmberg SD et al. Hepatitis C in the United States. N Engl J Med 2013;368:1859-1961
6. Di Bisceglie A et al., Virologic Outcomes and Adherence to Treatment Algorithms in a Longitudinal Study of Patients with Chronic Hepatitis C Treated with Boceprevir (BOC) or Telaprevir (TVR) in the United States (HCV-TARGET). 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; abstract 41
7. Fontaine H, et al., ANRS CO20 CUPIC study group. SVR12 rates and safety of triple therapy including Telaprevir or Boceprevir in 221 cirrhotic non-responders treated in the French early access program (ANRS CO20-CUPIC) 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 60
8. Rutter et al, Oral presentation, 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 65
9. Mauss S et al., Safety and efficacy of triple therapy containing boceprevir (BOC) or telaprevir (TVR) plus peginterferon alfa-2a/ ribavirin in patients with advanced fibrosis or cirrhosis in real-life setting. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; abstract 1856
10. Cotte L et al., High End-Of-Treatment (EOT) Response Rate with Telaprevir-PegIFN-RBV in Treatment-Experienced HIV Coinfected Patients with HCV genotype 1: ANRS HC26 TelapreVIH Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; abstract 1108
11. Poizot-Martin I et al., W48 Response Rate of Boceprevir-PegIFN-RBV in Treatment-Experienced HIV Coinfected Patients with HCV genotype 1: ANRS-HC27 BocepreVIH Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1105
12. Rockstroh K et al., STARTVerso 4 Phase III trial of faldaprevir plus peg interferon alfa-2a and ribavirin (PR) in patients with HIV and HCV genotype 1 coinfection: end of treatment response. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; abstract 1099
13. Sulkowski M et al., All-Oral Therapy With Sofosbuvir Plus Ribavirin For the Treatment of HCV Genotype 1, 2, and 3 Infection in Patients Co-infected With HIV (PHOTON-1). 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, SA; abstract 212
14. Cotte L,et al., and ANRS HC26 Study Group. High Early Virological Response with Telaprevir-Pegylated-Interferon-Ribavirin in Treatment-experienced Hepatitis C Virus Genotype 1/HIV Co-infected Patients: ANRS HC26 TelapreVIH Study. 20th Conference on Retroviruses and Opportunistic Infections, March 3-6, 2013; abstract 36
15. Poizot-Martin I, et al., and ANRS-HC27 BOCEPREVIH Study Group. ANRS-HC27 BocepreVIH Interim Analysis: High Early Virologic Response with Boceprevir + Pegylated Interferon + Ribivirin in Hepatitis C Virus/HIV Co-infected Patients with Previous Failure to Pegylated Interferon + Ribivirin. 20th Conference on Retroviruses and Opportunistic Infections, March 3-6, 2013; abstract 37
16. Montes M et al., Telaprevir combination therapy in treatment-naïve and experienced patients co-infected with HCV and HIV. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 38
17. Rockstroh K et al., STARTVerso 4 Phase III trial of faldaprevir plus peg interferon alfa-2a and ribavirin (PR) in patients with HIV and HCV genotype 1 coinfection: end of treatment response. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1099
18. Sulkowski M et al., All-Oral Therapy With Sofosbuvir Plus Ribavirin For the Treatment of HCV Genotype 1, 2, and 3 Infection in Patients Co-infected With HIV (PHOTON-1). 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington ,USA; abstract 212
19. Fontaine H, Hezode C, Dorival C, Larrey D, Zoulim F, de Ledinghen V Canva V, Alric L, Bourlie M, Pol S, Poynard T, Riachi1 G, Bernard PH, Raabe JJ, Gournay J, Metivier S, Pawlotsky JM, Samuel D, Barthe Y, Carrat F, Bronowicki JP, ANRS CO20 CUPIC study group. SVR12 rates and safety of triple therapy including Telaprevir or Boceprevir in 221 cirrhotic non-responders treated in the French early access program (ANRS CO20-CUPIC) 48th Annual Meeting of the European Association for the Study of the Liver, April 24-28, 2013, Amsterdam, Netherlands; abstract 60
20. Curry MP et al., Pretransplant Sofosbuvir and Ribavirin to Prevent Recurrence of HCV Infection after Liver Transplantation 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 213
21. Charlton MA et al., Sofosbuvir and Ribavirin for the Treatment of Established Recurrent Hepatitis C Infection After LiverTransplantation: Preliminary Results of a Prospective, Multicenter Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington,USA; abstract LB2
22. Faisal N et al., Protease inhibitor-based triple therapy is highly effective in liver transplant recipients with genotype 1 hepatitis C recurrence: A Canadian multicenter experience. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 63
23. Coilly A et al., Sustained Virological Response After Protease Inhibitor-based therapy For Hepatitis C Recurrence After Liver Transplantation: A Multicentric European Experience. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 216
24. Brown K et al., Twice-Daily Telaprevir in Combination with Peginterferon Alfa-2a/Ribavirin in Genotype 1 HCV Liver Transplant Recipients: Interim Week 16 Safety and Efficacy Results of the Prospective, Multicenter REFRESH Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB2
25. Jacobson I et al., Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in treatment-naïve patients: efficacy in difficult-to-treat patient sub-populations in the QUEST 1 and 2 phase III trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1122
26. Forns X et al., Simeprevir (TMC435) with peg-interferon α-2a/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: efficacy and safety in patient sub-populations in the PROMISE phase III tria64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1092
27. Lenz O et al., Resistance analyses of HCV isolates from patients treated with simeprevir in phase 2b/3 studies. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1101
28. Jensen DM et al., A pooled analysis of two randomized, double-blind placebo-controlled Phase III trials (STARTVerso1&2) of faldaprevir plus pegylated interferon alfa-2a and ribavirin in treatment-naïve patients with chronic hepatitis C genotype-1 infection. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1088
29. Jacobson I et al., STARTVerso3: A randomized, double-blind, placebo-controlled Phase III trial of faldaprevir in combination with pegylated interferon alfa-2a and ribavirin in treatment-experienced patients with chronic hepatitis C genotype-1 infection. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1100
30. Mangia A et al., Virologic Response Rates to Sofosbuvir-Containing Regimens Are Similar in Patients With and Without Traditional Negative Predictive Factors: A Retrospective Analysis of Phase 3 Data. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1115
31. Patel K et al., Efficacy and Safety of Sofosbuvir in Patients According to Fibrosis Stage: An Analysis of Phase 3 Data. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1093
32. Lawitz E et al., Sofosbuvir in Combination With PegIFN and Ribavirin for 12 Weeks Provides High SVR Rates in HCV-Infected Genotype 2 or 3 Treatment Experienced Patients with and without Compensated Cirrhosis: Results from the LONESTAR-2 Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB4
33. Zeuzem S et al., Sofosbuvir + Ribavirin for 12 or 24 Weeks for Patients with HCV Genotype 2 or 3: the VALENCE trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1085
34. Ruane P et al., Sofosbuvir plus Ribavirin in the Treatment of Chronic HCV Genotype 4 Infection in Patients of Egyptian Ancestry. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1090
35. Gane E et al., Efficacy and Safety of an Interferon-Free Regimen of MK-5172 +Ribavirin for 12 Weeks or 24 Weeks in Treatment Naive, Non-cirrhotic Subjects With HCV GT1 Infection: The C-SPIRIT Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1110
36. Jacobson I et al., SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naïve and prior null responder patients: The COSMOS study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB3
37. Dufour JF et al., Interferon-Free Treatment with Faldaprevir, Deleobuvir (BI 207127) and Ribavirin in SOUND-C3: 95% SVR12 in HCV-GT1b. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1102
38. Lalezari J et al., Rapid and Consistent Virologic Responses in a Phase 2 Trial of a New All-Oral Combination of Faldaprevir, Deleobuvir, and PPI-668, with and without Ribavirin, in Patients with HCV Genotype-1a Infection. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB20
39. Lawitz E et al., High Efficacy and Safety of the All-Oral Combination Regimen, MK-5172/MK-8742 +/- RBV for 12 Weeks in HCV Genotype 1 Infected Patients: The C-WORTHY Study. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, ; abstract 76
40. Gane E et al., Once Daily Sofosbuvir/Ledipasvir Fixed Dose Combination with or without Ribavirin: Data from the ELECTRON trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 73
41. Lawitz E et al., Once Daily Sofosbuvir/Ledipasvir Fixed Dose Combination with or without Ribavirin Resulted in ≥95% Sustained Virologic Response In Patients with HCV Genotype 1,Including Patients with Cirrhosis: the LONESTAR trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 215
42. Lawitz E et al., Once Daily Sofosbuvir/Ledipasvir Fixed Dose Combination is Highly Effective in Subjects with Baseline NS5A Inhibitor and NS3 Protease Inhibitor Resistance-Associated Variants: The Lonestar Trial 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1844
43. Cohen D et al., Safety of Ribavirin-containing Regimens of ABT-450/r, ABT-333, and ABT-267 for the Treatment of HCV Genotype 1 Infection and Efficacy in Subjects with Ribavirin Dose Reductions 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 1118
44. Chayama K et al., All-oral Combination of Daclatasvir Plus Asunaprevir in Interferon Ineligible Naive/Intolerant and Nonresponder Japanese Patients Chronically Infected with HCV Genotype 1b: Results from a Phase 3 Trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 211
45. Lawitz E et al., Interferon- and Ribavirin-free Regimen of ABT-450/r + ABT-267 in HCV Genotype 1b-infected Treatment-naïve Patients and Prior Null Responders. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract 75
46. Everson GT et al., Phase 2b study of the interferon-free and ribavirin-free combination of daclatasvir, asunaprevir, and BMS-791325 for 12 weeks in treatment-naïve patients with chronic HCV genotype 1 infection. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB1
47. Kohli A et al., Combination Oral, Ribavirin Free, Antiviral Therapy to Optimize Treatment Outcomes for Hepatitis C GT-1 Treatment Naïve Patients: Interim Results from the NIAID SYNERGY Trial. 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington, USA; abstract LB8
|
| |
|
|
|
|
|