| |
Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study
|
| |
| |
supplementary appendix attached
The Lancet, Early Online Publication, 3 July 2013
"At week 48, 251 (71%) patients on dolutegravir had HIV-1 RNA less than 50 copies per mL versus 230 (64%) patients on raltegravir (adjusted difference 7·4%, 95% CI 0·7 to 14·2); superiority of dolutegravir versus raltegravir was then concluded (p=0·03)......Overall, these data show that dolutegravir 50 mg given once daily combined with at least one active background drug provides superior virological control compared with raltegravir 400 mg given twice daily in ART-experienced, integrase-inhibitor-naive populations.......demonstration of potency is evidenced by the subgroups in which dolutegravir outperformed raltegravir, such as the more difficult-to-treat populations (eg, patients with baseline HIV RNA >50 000 copies per mL and patients not taking darunavir-ritonavir or taking it despite the presence of primary protease inhibitor resistance mutations)......Since SAILING was designed as a double-blind, double-dummy trial, one limitation of the study is that we could not assess the possible advantage of dolutegravir as a once-daily drug. In summary, the SAILING results confirm dolutegravir as an improved integrase-inhibitor-based treatment option for ART-experienced patients with multiclass resistance."
".....Current treatment guidelines for antiretroviral-experienced patients recommend that a failing regimen should be stopped as soon as possible to avoid progressive accumulation of resistance mutations"
"(see resistance review in Discussion section).......There was no emergence of primary or secondary raltegravir resistance mutations in the dolutegravir group.....treatment-emergent phenotypic resistance to dolutegravir or raltegravir was not reported in any patients on dolutegravir.......These data add to results from the SINGLE and SPRING-2 studies,14, 15 wherein no patients randomly assigned to dolutegravir have developed treatment-emergent resistance to dolutegravir or the nucleoside reverse transcriptase inhibitor (NRTI) backbone, while patients on efavirenz-tenofovir-emtricitabine (Atripla, Bristol-Myers Squibb Company, New York, NY, USA) or raltegravir plus two NRTIs, respectively, have developed resistance to both components.......treatment-emergent phenotypic resistance to dolutegravir or raltegravir was not reported in any patients on dolutegravir. Raltegravir-associated genotypic resistance9, 11 was recorded in 16 (42%) of 38 patients with protocol-defined virological failure in the raltegravir group, with high fold-change to raltegravir but limited cross-resistance to dolutegravir (appendix). 12 (3%) of 361 patients on raltegravir versus four (1%) of 354 on dolutegravir had treatment-emergent resistance to their background regimen at week 48 (appendix), a significant difference that was not included in our multiplicity-controlled testing procedure......2 (3%) of 361 patients on raltegravir versus four (1%) of 354 on dolutegravir had treatment-emergent resistance to their background regimen at week 48 (appendix), a significant difference......The emergent mutations in the raltegravir group of this study were, as previously reported, commonly observed, clinically relevant integrase inhibitor resistance mutations with associated decreases in raltegravir susceptibility. For dolutegravir, only two patients had unique integrase substitutions that were previously only identified in in-vitro passage studies with dolutegravir or elvitegravir but were not associated with clinically relevant decreased susceptibility to dolutegravir. Additionally, more patients on raltegravir developed resistance to background antiretroviral drugs"
"The favourable safety profile of dolutegravir after at least 48 weeks of treatment is consistent with previous reports for the integrase inhibitor class. Both dolutegravir-based and raltegravir-based therapy were well tolerated in this patient population, with few safety-related discontinuations. The types and frequency of adverse events for both treatment groups were as expected, based on the safety profile observed in a study comparing these drugs in treatment-naive patients.14 Changes in serum creatinine in dolutegravir recipients were consistent with previous findings and not regarded as clinically significant. Dolutegravir inhibits the organic cation transporter OCT2, similar to other drugs such as trimethoprim or cimetidine,26, 27 which decrease tubular secretion of creatinine and therefore increase concentrations of serum creatinine without affecting glomerular filtration"
"Rates of ALT increases greater than five times the upper limit of normal were also comparable between groups. In the patients who met liver stopping criteria, all but one event on raltegravir were assessed as related to concomitant disease (eg, hepatitis C or B virus, or both, with or without IRIS) or concomitant treatments. Importantly, patients co-infected with hepatitis B virus on dolutegravir who had significant liver chemistry increases were also not receiving active therapy for hepatitis B during the study, which, along with robust virological responses, might have triggered the ALT increases. Liver enzyme increases in patients co-infected with hepatitis C were more frequently of low grade and not treatment-limiting. Finally, a low and balanced incidence of other IRIS events adjudicated by the data monitoring committee in both groups of this study was observed."
Summary
Background
Dolutegravir (GSK1349572), a once-daily HIV integrase inhibitor, has shown potent antiviral response and a favourable safety profile. We evaluated safety, efficacy, and emergent resistance in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV-1 with at least two-class drug resistance.
Methods
ING111762 (SAILING) is a 48 week, phase 3, randomised, double-blind, active-controlled, non-inferiority study that began in October, 2010. Eligible patients had two consecutive plasma HIV-1 RNA assessments of 400 copies per mL or higher (unless >1000 copies per mL at screening), resistance to two or more classes of antiretroviral drugs, and had one to two fully active drugs for background therapy.
Participants were randomly assigned (1:1) to once-daily dolutegravir 50 mg or twice-daily raltegravir 400 mg, with investigator-selected background therapy. Matching placebo was given, and study sites were masked to treatment assignment.
The primary endpoint was the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL at week 48, evaluated in all participants randomly assigned to treatment groups who received at least one dose of study drug, excluding participants at one site with violations of good clinical practice. Non-inferiority was prespecified with a 12% margin; if non-inferiority was established, then superiority would be tested per a prespecified sequential testing procedure. A key prespecified secondary endpoint was the proportion of patients with treatment-emergent integrase-inhibitor resistance. The trial is registered at ClinicalTrials.gov, NCT01231516.
Findings
Analysis included 715 patients (354 dolutegravir; 361 raltegravir).
At week 48, 251 (71%) patients on dolutegravir had HIV-1 RNA less than 50 copies per mL versus 230 (64%) patients on raltegravir (adjusted difference 7·4%, 95% CI 0·7 to 14·2); superiority of dolutegravir versus raltegravir was then concluded (p=0·03).
Significantly fewer patients had virological failure with treatment-emergent integrase-inhibitor resistance on dolutegravir (four vs 17 patients; adjusted difference -3·7%, 95% CI -6·1 to -1·2; p=0·003).
Adverse event frequencies were similar across groups; the most commonly reported events for dolutegravir versus raltegravir were diarrhoea (71 [20%] vs 64 [18%] patients), upper respiratory tract infection (38 [11%] vs 29 [8%]), and headache (33 [9%] vs 31 [9%]).
Safety events leading to discontinuation were infrequent in both groups (nine [3%] dolutegravir, 14 [4%] raltegravir).
Interpretation
Once-daily dolutegravir, in combination with up to two other antiretroviral drugs, is well tolerated with greater virological effect compared with twice-daily raltegravir in this treatment-experienced patient group.
Funding
ViiV Healthcare.
Introduction
Although the first-generation integrase inhibitors raltegravir and elvitegravir are potent and well tolerated in treatment-naive and treatment-experienced adults with HIV,1-4 improved integrase inhibitor-based therapy options would benefit these patients. Raltegravir requires twice-daily dosing5 and has variable pharmacokinetic characteristics. Elvitegravir must be taken with food and requires coadministration with a pharmacokinetic boosting agent, creating the potential for clinically significant drug interactions.6 Additionally, treatment-experienced patients who fail raltegravir-based or elvitegravir-based regimens commonly develop integrase inhibitor resistance; cross-resistance between raltegravir and elvitegravir has been reported.4,7-11 Thus, the development of new unboosted integrase inhibitors with once-daily dosing and an improved resistance profile is desirable.
Dolutegravir (GSK1349572) is a next-generation integrase inhibitor in clinical development. Dolutegravir dissociates slowly from integrase-DNA complexes (t 1/2=71 h, wildtype integrase) in vitro,12 and has a 14-h plasma half-life in patients, supporting once-daily dosing without pharmacokinetic boosters.13 It has the potential for a high barrier to resistance and has shown potent efficacy in antiretroviral-naive patients and patients with multiclass resistance.14-16 No significant food effect or significant cytochrome P450 inhibition or induction has been observed, suggesting a low potential for interactions.13, 17
We report results from ING111762 (SAILING), comparing clinical efficacy, safety, and virology outcomes in treatment-experienced, integrase-inhibitor-naive patients who received dolutegravir 50 mg once daily or raltegravir 400 mg twice a day, plus investigator-selected background therapy.
see Methods below
Results
Of 1441 patients screened, 724 were randomly assigned to treatment groups and 719 received at least one dose of study drug (dolutegravir, 357; raltegravir, 362; figure 1). Baseline demographics, disease characteristics, treatment history, and background drugs and their activity were balanced across treatment groups (table 1). Because of broad geographical participation, the SAILING population was diverse in ethnicity, sex, and HIV-1 subtype (B [487 patients, 68%], C [103, 14%], and complex [42, 6%]), and generally had more advanced disease (almost half had a history of AIDS), plus nearly half with resistance to at least one drug in each of three or more antiretroviral drug classes (table 1).
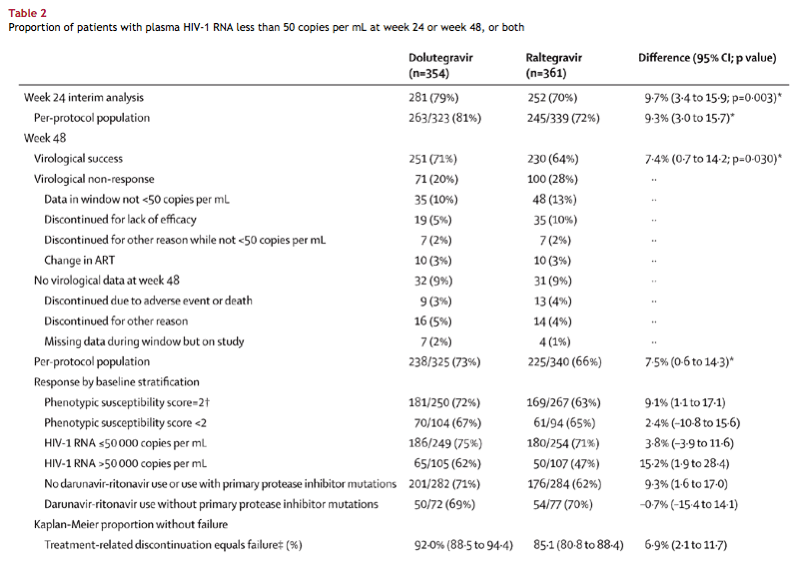
In both treatment groups, the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL increased sharply from baseline to week 4, then generally remained stable from week 8 onward (figure 2). At week 48, 251 (71%) of 354 patients receiving dolutegravir and 230 (64%) of 361 patients receiving raltegravir had plasma HIV-1 RNA lower than 50 copies per mL (table 2). Because the adjusted treatment difference (7·4%, 95% CI 0·7-14·2) was greater than -12%, dolutegravir was concluded to be non-inferior to raltegravir. Statistical superiority (p=0·03) was subsequently shown as part of a prespecified testing procedure. The difference in response rates between the two groups was mainly driven by virological outcomes.
Consistent with the primary analysis, per-protocol analyses and Kaplan-Meier estimates of the proportion of patients without treatment-related failure by week 48 indicated significant differences in favour of dolutegravir (table 2). For the subgroup of patients without primary protease inhibitor mutations taking darunavir-ritonavir, dolutegravir and raltegravir response rates were similar. Treatment differences within other subgroups (by baseline stratification) were generally supportive of the primary result (table 2).
from Jules: You can see in Table 2 immediately below for patients not taking darunavir or use with primary protease inhibitor mutations the proportion of patients with plasma HIV-RNA <50 c/ml is 72% with dolutegravir s 62% with raltegravir.
CD4+ cell counts increased from baseline to week 48 in both groups (mean change 162 [SD 151] cells per μL in the dolutegravir group; 153 [144] cells per μL on raltegravir). Protocol-defined virological failure occurred earlier and more frequently in the raltegravir group (45 [12%] patients vs 21 [6%] in the dolutegravir group by week 48). 19 (42%) of the 45 patients on raltegravir with virological failure were virological non-responders (as opposed to virological rebounders) compared with two (10%) of 21 patients on dolutegravir.
Significantly fewer patients in the dolutegravir group had treatment-emergent genotypic or phenotypic evidence of integrase inhibitor resistance by week 48 (four [1%] of 354 vs 17 [5%] of 361 patients, p=0·003; adjusted difference -3·7%, 95% CI -6·1 to -1·2, p=0·003); of these, one patient in each group had raltegravir primary resistance at baseline (dolutegravir, Gln148His/Gly140Ser pathway; raltegravir, Tyr143 pathway). A unique integrase substitution (Arg263Lys or Arg263Arg/Lys mixture) was noted in two patients on dolutegravir (one clade B and one clade C virus) with protocol-defined virological failure that conferred a less than two fold-change in susceptibility to dolutegravir and raltegravir; one patient on dolutegravir with HIV-1 RNA greater than 90 000 copies per mL at all timepoints through virological failure developed a polymorphic Val151Val/Ile change that conferred no dolutegravir or raltegravir fold-change increase (appendix). Thus, treatment-emergent phenotypic resistance to dolutegravir or raltegravir was not reported in any patients on dolutegravir. Raltegravir-associated genotypic resistance9, 11 was recorded in 16 (42%) of 38 patients with protocol-defined virological failure in the raltegravir group, with high fold-change to raltegravir but limited cross-resistance to dolutegravir (appendix). 12 (3%) of 361 patients on raltegravir versus four (1%) of 354 on dolutegravir had treatment-emergent resistance to their background regimen at week 48 (appendix), a significant difference that was not included in our multiplicity-controlled testing procedure.
The safety profiles for dolutegravir and raltegravir were comparable, with similar rates of adverse events (table 3) and most events in both treatment groups were of mild to moderate intensity. The most commonly reported clinical adverse events for dolutegravir versus raltegravir groups were diarrhoea, upper respiratory tract infection, and headache (table 3). Safety events leading to discontinuation (adverse events or stopping criteria) were infrequent for both treatment groups (table 3). The rates and nature of serious adverse events were also similar, and few patients developed a drug-related serious adverse event (table 3). No deaths occurred in the dolutegravir group; three deaths occurred in the raltegravir group (metastatic adenocarcinoma, multiorgan failure, and cervical carcinoma; all judged unrelated to study drug).
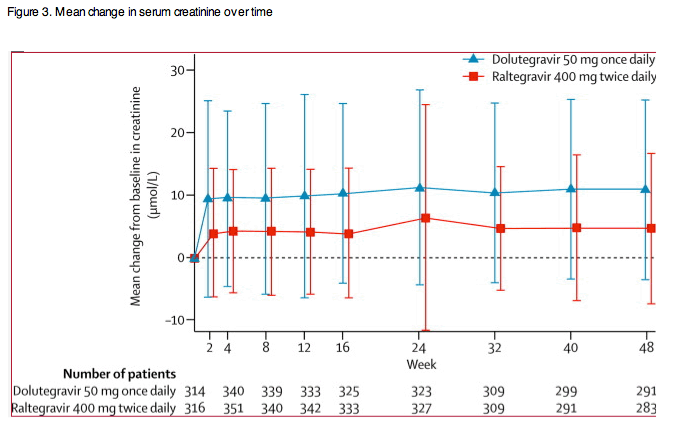
The distribution and number of graded treatment-emergent laboratory toxicities were similar between groups (table 3). Small increases in serum creatinine were evident in both groups at week 2 and remained stable to week 48 (figure 3). Few patients had grade 2 (dolutegravir, five; raltegravir, six) or grade 3 (one each) serum creatinine increases; these were mainly attributable to pre-existing renal conditions, hypertension, diabetes, drugs, or infectious complications (except one patient per group had transient grade 2 increases not attributable to other causes, though both continued on study drug). A small median reduction from baseline for urine albumin:creatinine ratios was noted in both groups (dolutegravir, -0·10 mg/mmol, IQR -0·85 to 0·20; raltegravir, -0·10 mg/mmol, -1·00 to 0·20).
Each group had a similar proportion of treatment-emergent increases in alanine aminotransferase (ALT) five or more times the upper limit of normal (nine [3%] of 357 patients on dolutegravir; seven [2%] of 362 on raltegravir). Seven patients on dolutegravir and four on raltegravir met protocol-defined stopping criteria for liver chemistry; causative diagnoses were adjudicated by the independent data monitoring committee (appendix). Patients with hepatitis B or C viruses were more likely to have immune reconstitution inflammatory syndrome (IRIS) or hepatic flares on dolutegravir in conjunction with improved virological and immunological responses, particularly when hepatitis B therapy was withdrawn at the start of the study or not coadministered. Five patients on dolutegravir and one patient on raltegravir were identified by the data monitoring committee as having hepatitis B or hepatitis C (or both) IRIS (one patient on dolutegravir and two on raltegravir had other IRIS events); other hepatitis diagnoses included alcohol intoxication, drug-induced liver injury (one related to raltegravir, one related to tipranavir-ritonavir for a patient on dolutegravir), acute infection, and gallstones. One patient on dolutegravir with active therapy for hepatitis B was withdrawn at day 1, having met liver stopping criteria because of hepatitis B virus IRIS, and was allowed to restart dolutegravir after addition of entecavir and hepatitis resolution. Liver enzyme increases did not recur after restart of dolutegravir. Similar low rates of post-baseline HIV-associated conditions (20 [6%] patients on dolutegravir, 19 [5%] on raltegravir, excluding recurrences) and post-baseline disease progression (progression to CDC class C or death; ten [3%] patients on dolutegravir, eight [2%] on raltegravir) were observed in both treatment groups up to week 48 (detailed data not shown). Similar minor improvements in utility values and modest gains in perceived health status (visual analogue score) occurred for both groups (data not shown).
Discussion
SAILING is the first study to show superior virological efficacy of any antiretroviral drug over raltegravir (panel, figure 2). This difference was driven by fewer virological non-responders in the dolutegravir group compared with the raltegravir group. Additionally, there was significantly less treatment-emergent integrase inhibitor or background therapy resistance at failure.
-------------------------------------
Panel
Research in context
Systematic review
We did a systematic search to address a clearly defined question: "Has any drug been shown superior to raltegravir in phase 3 clinical studies?" Search terms included "raltegravir AND Phase 3", "raltegravir AND versus phase 3". 30 articles were retrieved and hand-searched; of these, only two compared raltegravir with another drug in non-inferiority trials, indicating that the present study is the first to show superiority. Regimens that provide treatment-experienced individuals with HIV-1 with more convenient once-daily options with fewer treatment-limiting side-effects are important to improve adherence and re-establish virological suppression. Guideline experts have suggested that although virological failure in a patient can occur for many reasons, much virological failure on first-line regimens is due to suboptimum adherence. Current treatment guidelines for antiretroviral-experienced patients recommend that a failing regimen should be stopped as soon as possible to avoid progressive accumulation of resistance mutations. The guidelines also recommend that the new regimen should be optimised to contain at least two, and preferably three, fully active drugs. A fully active drug is one that is likely to have antiretroviral activity on the basis of the patient's treatment history, drug-resistance testing, or a novel mechanism of action. The integrase class has provided the opportunity to construct potent and well tolerated regimens.
Interpretation
This double-blind, active-controlled, phase 3 study compared the two integrase strand transfer inhibitors raltegravir and dolutegravir in combination with up to two background antiretroviral drugs in antiretroviral therapy-experienced, integrase-inhibitor-naive adults with at least two-class drug resistance. This study is the first to show higher virological potency of any antiretroviral drug over raltegravir. Both integrase-inhibitor-containing regimens were well tolerated with infrequent discontinuations related to adverse events. The emergent mutations in the raltegravir group of this study were, as previously reported, commonly observed, clinically relevant integrase inhibitor resistance mutations with associated decreases in raltegravir susceptibility. For dolutegravir, only two patients had unique integrase substitutions that were previously only identified in in-vitro passage studies with dolutegravir or elvitegravir but were not associated with clinically relevant decreased susceptibility to dolutegravir. Additionally, more patients on raltegravir developed resistance to background antiretroviral drugs. Taken together, the results of this head-to-head phase 3 study suggest that once-daily dolutegravir 50 mg, in combination with up to two other antiretroviral drugs, is well tolerated and effective as therapy for antiretroviral treatment-experienced, integrase-inhibitor-naive adults and is an alternative to twice-daily raltegravir.
---------------------------------------------
Data from SAILING show that dolutegravir in combination with up to two additional ARTs has higher virological efficacy and a higher barrier to resistance compared with raltegravir, as supported by two prespecified analyses. The proportion of patients harbouring virus with evidence of treatment-emergent genotypic or phenotypic integrase inhibitor resistance was greater for raltegravir versus dolutegravir; the proportion of patients with resistance to the background regimen was also greater for the raltegravir group. There was no emergence of primary or secondary raltegravir resistance mutations in the dolutegravir group. Instead, one patient developed the polymorphic integrase substitution Val151Val/Ile with no increased dolutegravir or raltegravir fold change, and two patients developed unique Arg263Lys or Arg263Arg/Lys substitutions (one clade B and one clade C virus), both with dolutegravir or raltegravir fold change of less than two, suggesting no high-level resistance to either drug is conferred by the Arg263Lys substitution. Of note, the Arg263Lys substitution has been selected during in-vitro passage with elvitegravir and dolutegravir.24, 25 The emergent mutations in the raltegravir group of this study were commonly observed, clinically relevant integrase inhibitor resistance mutations.9 These data add to results from the SINGLE and SPRING-2 studies,14, 15 wherein no patients randomly assigned to dolutegravir have developed treatment-emergent resistance to dolutegravir or the nucleoside reverse transcriptase inhibitor (NRTI) backbone, while patients on efavirenz-tenofovir-emtricitabine (Atripla, Bristol-Myers Squibb Company, New York, NY, USA) or raltegravir plus two NRTIs, respectively, have developed resistance to both components. Protocol-defined virological failure occurred earlier and more frequently in the raltegravir group, reflecting the greater antiviral potency of dolutegravir in this patient population. Further demonstration of potency is evidenced by the subgroups in which dolutegravir outperformed raltegravir, such as the more difficult-to-treat populations (eg, patients with baseline HIV RNA >50 000 copies per mL and patients not taking darunavir-ritonavir or taking it despite the presence of primary protease inhibitor resistance mutations).
The favourable safety profile of dolutegravir after at least 48 weeks of treatment is consistent with previous reports for the integrase inhibitor class. Both dolutegravir-based and raltegravir-based therapy were well tolerated in this patient population, with few safety-related discontinuations. The types and frequency of adverse events for both treatment groups were as expected, based on the safety profile observed in a study comparing these drugs in treatment-naive patients.14 Changes in serum creatinine in dolutegravir recipients were consistent with previous findings and not regarded as clinically significant. Dolutegravir inhibits the organic cation transporter OCT2, similar to other drugs such as trimethoprim or cimetidine,26, 27 which decrease tubular secretion of creatinine and therefore increase concentrations of serum creatinine without affecting glomerular filtration.28, 29 Rates of ALT increases greater than five times the upper limit of normal were also comparable between groups. In the patients who met liver stopping criteria, all but one event on raltegravir were assessed as related to concomitant disease (eg, hepatitis C or B virus, or both, with or without IRIS) or concomitant treatments. Importantly, patients co-infected with hepatitis B virus on dolutegravir who had significant liver chemistry increases were also not receiving active therapy for hepatitis B during the study, which, along with robust virological responses, might have triggered the ALT increases. Liver enzyme increases in patients co-infected with hepatitis C were more frequently of low grade and not treatment-limiting. Finally, a low and balanced incidence of other IRIS events adjudicated by the data monitoring committee in both groups of this study was observed.
Overall, these data show that dolutegravir 50 mg given once daily combined with at least one active background drug provides superior virological control compared with raltegravir 400 mg given twice daily in ART-experienced, integrase-inhibitor-naive populations. Since SAILING was designed as a double-blind, double-dummy trial, one limitation of the study is that we could not assess the possible advantage of dolutegravir as a once-daily drug. In summary, the SAILING results confirm dolutegravir as an improved integrase-inhibitor-based treatment option for ART-experienced patients with multiclass resistance.
Methods
Study design and patients
SAILING is an ongoing phase 3, randomised, double-blind, active-controlled, double-placebo, multicentre, parallel-group, non-inferiority study in treatment-experienced, integrase-inhibitor-naive, adults with HIV-1 at 156 centres in Australia, Canada, Europe, Latin America, Taiwan, South Africa, and the USA (appendix). The screening period was Oct 26, 2010, to Jan 20, 2012. Eligible participants had two consecutive plasma HIV-1 RNA assessments of 400 copies per mL or higher (unless >1000 copies per mL at screening), resistance to two or more classes of antiretroviral drugs, and had one to two fully active agents for background therapy. Exclusions included active US Centers for Disease Control and Prevention category C disease18 (except Kaposi's sarcoma), defined laboratory values, pregnancy, moderate or severe hepatic impairment, expected need for hepatitis C virus therapy, malignancy, or recent treatment with HIV-1 vaccines, radiation therapy, cytotoxic chemotherapy, or immunomodulators (full eligibility criteria in appendix).
Patients randomly assigned to treatment groups received dolutegravir 50 mg once a day or raltegravir 400 mg twice a day plus investigator-selected background therapy (at least one fully active agent with or without a second agent, with or without full activity). Planned use of darunavir-ritonavir without primary protease inhibitor resistance on screening genotype was a stratification factor (see below), and was to be capped at 170 to help to delineate dolutegravir contributions to virological suppression.
Ethics committee approval was obtained at all participating centres in accordance with the principles of the 2008 Declaration of Helsinki. Each patient provided written informed consent before undergoing study procedures.
Randomisation and masking
Central, computer-generated randomisation (1:1) included stratification by HIV-1 RNA (≤50 000 vs >50 000 copies per mL); darunavir-ritonavir use without primary protease inhibitor resistance versus no use or use with primary protease inhibitor mutations; and two versus fewer than two fully active background agents. Up to week 48, matching placebo was given with masked dolutegravir or raltegravir, and all study sites remained masked. A week 24 interim analysis was done to enable regulatory submission of dolutegravir; sponsor staff had access to data from this point forward.
Procedures
Clinical and laboratory analyses occurred at baseline and weeks 2, 4, 8, 12, 16, 24, 32, 40, and 48. After successful completion of week 48, patients randomly assigned to dolutegravir have open-label access to dolutegravir and are followed up every 12 weeks thereafter.
Central laboratory facilities (Quest Diagnostics, Valencia, CA, USA) provided haematology, clinical chemistry, urinalysis, and plasma HIV-1 RNA testing (with the RealTime HIV-1 PCR assay [Abbott Molecular, Des Plaines, IL, USA]). CD4+ cell count and percentage were measured at each visit (except week 2). Monogram Biosciences (San Francisco, CA, USA) did integrase inhibitor resistance testing for day 1 and protocol-defined virological failure samples using PhenoSense Integrase and Geneseq Integrase, and used PhenoSense GT, PhenoSense Entry, and Trofile assays for other classes.
The primary endpoint was the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL at week 48. A key secondary endpoint was the proportion of patients with treatment-emergent integrase inhibitor resistance. Other prespecified efficacy endpoints included the proportion of patients with evidence of treatment-emergent resistance to the background regimen, changes from baseline in CD4 cell counts, the incidence of HIV-associated conditions, and incidence of disease progressions (HIV-associated conditions, AIDS, and death). The main safety endpoints were the incidence and severity of adverse events and changes in laboratory parameters. Other secondary endpoints were utility and visual analogue scale measures of quality of life using the EQ-5D health questionnaire (EuroQol, Rotterdam, Netherlands), dolutegravir pharmacokinetic characteristics, and pharmacokinetic and pharmacodynamic relations (data not shown). Adverse events were evaluated and graded according to the Division of AIDS toxicity scales.19 Liver chemistry threshold stopping criteria were implemented to assure patient safety and evaluate cause of liver inflammation.
Genotypic and phenotypic susceptibility scores were established with selected background drugs together with resistance results. Genotypic susceptibility score was calculated with the Stanford HIVdb algorithm, July 2012 version20 (appendix). Phenotypic susceptibility score was based on Monogram Biosciences phenotypic resistance testing on the baseline sample; a score of 1 was assigned if fold change was above the lower clinical cutoff or biological cutoff, and a score of 1 was assigned for maraviroc when CXCR4-tropic HIV-1 was not detected. For the purpose of statistical analysis, if no resistance results were available at baseline to confirm the activity of a background drug, then a susceptibility score of 0 was assumed. Integrase inhibitor resistance testing was not routinely done on baseline samples in the absence of protocol-defined virological failure because of the low prevalence of transmitted integrase inhibitor resistance in patients who are naive to these drugs.
Resistance testing (genotype and phenotype) was done on samples for which the following conditions were met and confirmed for protocol-defined virological failure: virological non-response (plasma HIV-1 RNA decrease <1 log10 copies per mL unless <400 copies per mL by week 16 or HIV-1 RNA ≥400 copies per mL on or after week 24) or virological rebound (plasma HIV-1 RNA ≥400 copies per mL after confirmed HIV-1 RNA <400 copies per mL or >1 log10 copies per mL increase above any nadir of ≥400 copies per mL). Patients with confirmed protocol-defined virological failure were with-drawn from the study.
Statistical analysis
The primary efficacy hypothesis was that dolutegravir would have non-inferior antiviral activity to that of raltegravir at week 48, when both are given with background therapy. Non-inferiority would be concluded if the lower bound of a two-sided 95% CI for the difference in proportions (dolutegravir minus raltegravir) of patients with plasma HIV-1 RNA less than 50 copies per mL at week 48 was greater than -12%. This prespecified non-inferiority margin was based on the observed benefit of raltegravir versus placebo for patients with background regimen phenotypic susceptibility scores of 1 and 2 in the BENCHMRK studies,3, 9 and on applicable HIV-specific21 and statistical guidelines.22
On the assumption of a 65% response rate in the raltegravir group, the study needed 333 evaluable participants per group to have 90% power with a one-sided 2·5% significance level. The primary analysis included all participants randomly assigned to treatment groups who received at least one dose of study drug, excluding four participants at one site with violations of good clinical practice. Per-protocol analyses excluded patients with prespecified protocol deviations. If non-inferiority was established in both primary and per-protocol analyses, then superiority would be tested at the nominal 5% significance level.
For the primary analysis, antiviral response was assessed with the snapshot algorithm defined by the US Food and Drug Administration.23 Patients whose last HIV-1 RNA result was less than 50 copies per mL in the analysis window (week 48, within 6 weeks) were regarded as responders; non-responders were patients with unsuppressed viral load or who withdrew without viral load data at the analysis timepoint. The protocol allowed one within-class switch to the background regimen for management of toxic effects; patients who switched antiretroviral therapy (ART) after week 4 were regarded as non-responders according to the snapshot algorithm. The difference in the proportions was adjusted for randomisation stratification factors (using baseline values).
Secondary efficacy analyses done to support the primary analyses included Kaplan-Meier estimates for the proportion of patients without virological failure by week 48, using treatment-related discontinuation equal failure analysis as previously described.14
The secondary endpoint (prespecified and α-controlled) of proportion of patients with evidence of treatment-emergent genotypic or phenotypic integrase inhibitor resistance at the time of protocol-defined virological failure was compared with a Cochran-Mantel-Haenszel analysis. The type I error rate was controlled for this secondary endpoint with a prespecified fixed-sequence testing procedure that allowed for secondary endpoint testing at the nominal 5% level if non-inferiority was shown for the primary endpoint. To assess treatment-emergent resistance to the background regimen during protocol-defined virological failure, the proportions of patients with evidence of a decrease in background regimen genotypic and phenotypic susceptibility scores from baseline to protocol-defined virological failure were compared with Cochran-Mantel-Haenszel statistics in a prespecified analysis.
This study is registered at ClinicalTrials.gov, NCT01231516.
Role of the funding source
The study was sponsored by ViiV Healthcare. All operational aspects of the study, including monitoring, data collection, and statistical analyses, were managed by GlaxoSmithKline. All authors had full access to all the study data and are responsible for the veracity and completeness of the data reported. The corresponding author had final responsibility for the decision to submit for publication.
| |
| |
| |
|
|
|