 |
|
|
| |
Summary from CROI 2014 for Hepatitis
HCV direct acting antivirals (DAAs) in HCV and HIV/HCV coinfection: same outcome?
Which DAA regime will allow for the shortest treatment duration?
|
| |
| |
Jurgen K. Rockstroh M.D., Professor of Medicine
University of Bonn, Germany
Introduction
At this year CROI viral hepatitis has clearly been one of the major topics with many exciting reports on treatment of hepatitis C in coinfection but also increasingly for hepatitis C mono-infection. The data presented covers all aspects of viral hepatitis including natural history, pathogenesis of disease, new fibrosis markers in liver disease, treatment trials and finally also some pharmacology results from PK work around HCV drugs and their potential for drug-drug interactions. Looking at the SVR rates of the recent coinfection trials which were presented at CROI 2014 in Boston - including triple HCV therapy with PEG-IFN/RBV and any first or second wave HCV protease inhibitor such as telaprevir, boceprevir or faldaprevir or simeprevir as well as the results from the first IFN-free study (PHOTON-1) - it becomes evident that HCV cure rates in the era of successful combination antiretroviral therapy (cART) are completely comparable to HCV SVR rates in HIV mono-infected patients (1-6). Therefore it is time to stop regarding coinfected patients as a separate risk group, which is more difficult to treat but rather demand the inclusion of dually HIV/HCV coinfected subjects into HCV trials irrespective of their HIV infection. Only exception is the need to consider potential drug-drug interactions between HIV and HCV drugs. Indeed in the ongoing development trial plan of the Merck IFN-free HCV regimen HIV/HCV coinfected patients were allowed to enter an ongoing HCV trial which is the first time that this specific patient group was allowed into a previously HCV mono-infection trial rather than being enrolled into a separate coinfection trial (7). Hopefully, the comparable cure rates in coinfection will help to decrease the barriers in HCV treatment uptake in coinfection and lead to a more wide-spread use and access of new HCV therapies in this patient group.
Efficacy and safety of telaprevir or boceprevir in HIV/HCV coinfection
New data on efficacy and safety of telaprevir or boceprevir in HIV/HCV coinfected previous non-responders to HCV therapy
In the poster hepatitis session final results from two ANRS studies were presented which looked at the efficacy and safety of triple HCV therapy (with either telaprevir or boceprevir) in previous non-responders to dual PEG-IFN/RBV therapy (last CROI interim results of these two trials were presented for the first time). Patients with previous null-response and cirrhosis however, were excluded because of the overall low probability of treatment response. In the telaprevir ANRS HC26 study overall 69 patients who received at least one dosage were included (1). Background HIV therapy contained ATV, ATV/r, EFV, RAL, TDF, FTC, or 3TC. The study design is depicted below (figure 1):
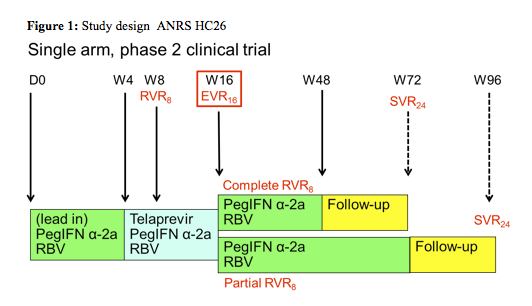
Patients started with a 4 week lead-in of PEG-IFN/RBV. This approach, which is outside the labelling of telaprevir, was chosen to be able to compare the results to the study design wise similar boceprevir study and because data from telaprevir studies in HCV mono-infection in treatment experienced patients had potentially promised some benefit for this approach. After week 4 telaprevir was added for12 weeks. Patients who achieved a complete rapid virological response at week 8 (RVR8) defined as HCV-RNA <15 IU/mL received 32 weeks of PEG-IFN/RBV after stopping telaprevir at week 16 after 12 weeks of triple therapy (full treatment: 48 weeks). Whereas patients who only obtained a partial RVR8 (15 IU/mL < HCV-RNA < 1000 IU/mL) received an additional 56 weeks of PEG-IFN/RBV (full treatment: 72 weeks). Telaprevir was administered as 750 mg q8h (1125 mg q8h with Efavirenz) and Peg-IFN α-2a as 180 μg sc / week. Ribavirin was dosed as 1000 mg / day (≤75 kg) and 1200 mg /day (>75 kg). EPO, G-CSF, and TPO-R agonists were allowed within the study. Futility rules for telaprevir were HCV-RNA > 1000 IU/mL at week 8 or week 12 or virological breakthrough at any time. For study inclusion patients needed to have CD4 ≥ 200 cells/mm³ and ≥ 15%, as well as plasma HIV-RNA levels < 50 copies/mL At baseline median CD4-count was 630 cells/mm³ (range 459-736) and HIV-RNA was below 50 copies in 99% of patients. Interestingly, 11 (18%) patients had F3 fibrosis and 16 (23%) F4 fibrosis upon inclusion to the study, allowing studying safety and efficacy of telaprevir based triple therapy in this more difficult-to-treat patient population. 39% of patients were previous relapsers to dual therapy, 9% had previous viral breakthrough, 22% were partial responders and 30% null-responders.
The final efficacy results are presented below:
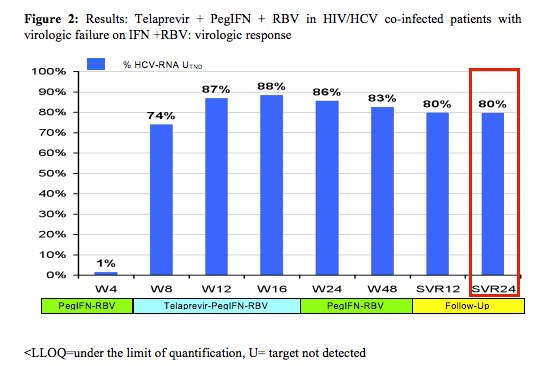
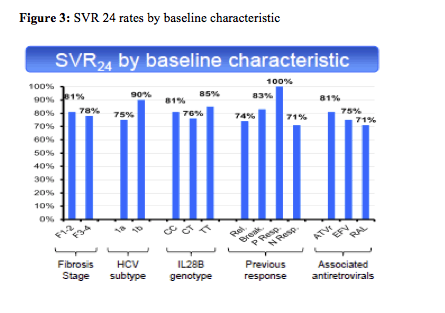
With 80% remaining HCV-RNA negative 24 weeks after stopping HCV therapy in this patient population with 30% previous null-responders and 40% F3/F4 fibrosis this study shows surprising high efficacy results. Most interestingly efficacy remained high throughout all sub analyses (see figure 3).
10% (n=7) of patients discontinued study drugs because of adverse events prior to week 16, 10% thereafter. 16% developed Grade 4 hematological adverse events. The authors concluded that significant hematological toxicity was observed, despite a proactive management of anemia. This is very well in line with results from the CUPIC trial (a HCV-PI + PEG-IFN/RBV trial in HCV monoinfected in patients with CHILD A cirrhosis) which also had reported higher anemia rates in this patient population with more advanced fibrosis. Further insights into the anemia rate associated with telaprevir came from additional analyses from this study as a late breaker poster (8). In this particular analyses the relationship between ribavirin (RBV) trough concentration (C), estimated glomerular filtration rate (eGFR) and hemoglobin (Hb) level, was examined before and after introduction of TVR in the TelapreVIH study. Interestingly, the addition of TVR to PegIFN+RBV was associated with a decrease in eGFR and a significant increase in RBV concentrations, leading to RBV overexposure in 45% of the patients explaining the high haematological toxicity observed in the trial.
The second ANRS study looked at the efficacy and safety of a boceprevir containing triple therapy again in previous IFN/RBV non-responders (n=64) (2). The study design is depicted below:
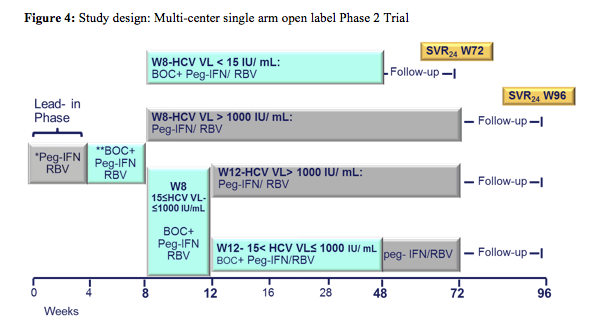
Following a lead-in with dual therapy boceprevir was added at week 4. Complex futility rules were in place during the study warranting discontinuation of boceprevir in all patients with a HCV viral load above 1000 IU/ml at week 8 as well as in week 12. All HCV drugs were discontinued if HCV viral load was >1000 IU/ml at week 16 or still detectable at week 28 and in case of virological breakthrough. Duration of therapy was dependent on initial response as shown above. Patients who achieved a rapid virological response and had an HCV-RNA <15 IU/ml at week 8 were treated for 48 weeks; patients with slower decline in HCV RNA were treated for 72 weeks. Patients recruited for the trial had to be on stable ART for at least 3 months, with at least three molecules among ATV (rtv boosted or not), RAL, TDF, ABC, FTC, or 3TC. Again only previous non-responders to dual therapy with HCV genotype 1 infection were included. Null-responders with cirrhosis were excluded from this trial. Overall, 17% of patients at baseline had F4 fibrosis, 33% were previous null-responders. The corresponding response rate is depicted below (figure 5):
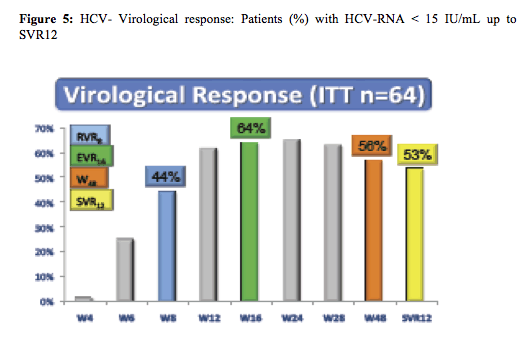
The overall cure rates look somewhat lower than what was achieved in the Telaprevir based study presented above. Indeed further sub analyses of overall response rates by baseline risk factors depicts particularly low response rates in previous non-responders (see also figure 6).
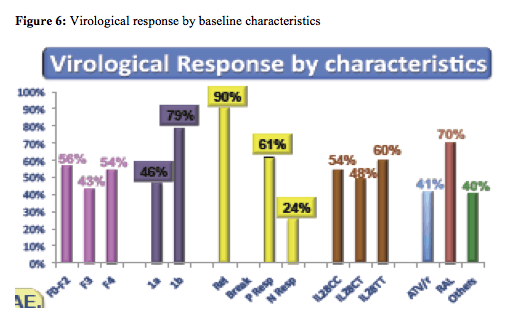
Clearly the overall small number of study subjects limits the strength of the comparisons. The safety data again suggests higher haematological adverse events than in the treatment naïve patient population with less fibrosis. 38/64 patients received concomitant EPO, 8 received transfusions. Overall discontinuation rate because of adverse events was 10%.
In summary, these first two studies in previous non-responders to dual therapy with PEG-IFN/RBV and a considerable proportion of patients with more advanced fibrosis stages demonstrate much higher cure rates than previously seen in HIV/HCV coinfection under dual therapy. In particular in the telaprevir trial very high cure rates of 80% were achieved. This data is particularly important for those countries were first generation HCV protease inhibitors have just become available and were it potentially still can take quite some time before other more recent DAAs will become registered and fully reimbursed. These data obtained in much more challenging to treat patients suggest that even in treatment experienced patients (only exception cirrhotic previous non-responders were not included so data for that specific patient group) good chances for HCV cure exist. Treatment with these first HCV triple therapies however, comes at the burden of increased toxicities and significant pill burden as well as still considerably long treatment durations highlighting why more effective and safer regimens are needed as fast as possible.
Further data on use of telaprevir and boceprevir in the clinics outside of clinical trials
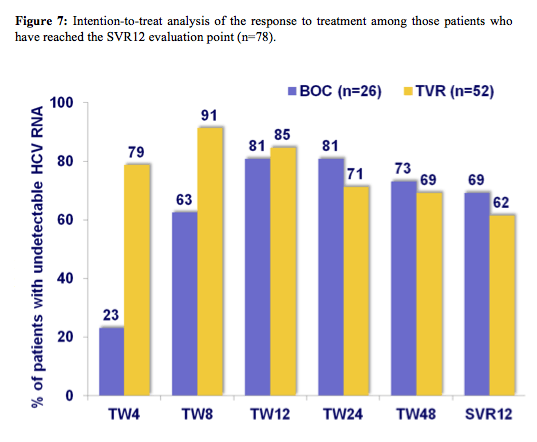
In addition to the ANRS presentations results from the use of boceprevir and telaprevir from hepatitis clinics in Germany, Spain, and Switzerland were presented (3, 9). Overall, low uptake of these new HCV triple therapies were reported reflecting the concerns around treatment related toxicities, high pill burden and potentially much easier treatment regimens to become available in the very near future. Nevertheless, it has to be stressed that in these real-life patient population a surprisingly high efficacy (see figure 7) was recorded considering that most patients at baseline had cirrhosis and a significant number of patients were prior non-responders. The overall SVR12 in this international cohort was 64.1% (50/78).
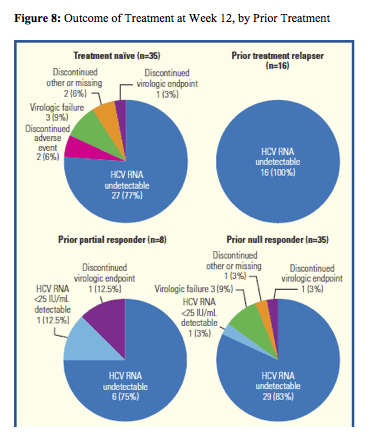
Interestingly, a first interim analysis of the HEP30005 study was presented which evaluates Telaprevir Treatment of HIV/HCV G1 Patients with Severe Fibrosis (10). This multicenter, open-label, early access program of telaprevir in combination with PR enrolled HIV/HCV coinfected subjects with HCV genotype 1 infection, severe fibrosis (F3) or compensated cirrhosis (F4). Overall, 118 patients are being treated with telaprevir plus PR for 12 weeks, followed by PR alone for 36 weeks. Data to Week 16 available for 102 patients were included in the interim analysis. Figure 8 summarizes the rate of undetectability at week 12 by prior response to HCV dual therapy.
Second-wave HCV protease inhibitors in HIV/HCV coinfection: final results
The next exciting presentations were around the use of 2nd wave HCV protease inhibitors in combination with PEG-IFN/RBV in HCV coinfected HIV-patients. Obviously the new HCV PIs which can be administered once daily and appear to have a more favorable adverse event profile promise a simplified and potentially better tolerated HCV therapy for coinfected subjects. In addition these new compounds show considerable genotype 4 activity promosing also potentially new treatment options for this increasing group of patients.
The first study evaluated the efficacy and safety of the HCV protease inhibitor faldaprevir (FDV) in combination with PEG-IFN and ribavirin in either treatment-naïve or HCV relapsers with GT1 infection and stable HIV coinfection. The study design of the FDV coinfection study is depicted below (4) - and there were several studies of the BI HCV protease inhibitor and its pharmacokinetic interactions with darunavir/ritonavir, efavirenz, and tenofovir with the HCV protease inhibitor faldaprevir (FDV) in healthy volunteers (9).
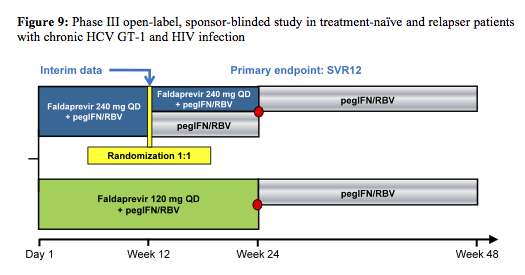
The red dot resembles patients with HCV RNA below LLoQ, at Week 4, and HCV RNA below LLoQ target not detected at Week 8 (=ETS) which were re-randomized 1:1 at week 24 to stop treatment or continue pegIFN/RBV through week 48. Patients who did not achieve ETS had to continue pegIFN/RBV through week 48. Therefore, this was one of the first coinfection studies to address the possibility of RVR guided shorter treatment durations. Patients with no ART or on maraviroc or Raltegravir were randomized to 120 or 240mg FDV QD, respectively. Because of previously described pk-interactions (CROI 2013) patients on efavirenz were allocated to 240mg QD and patients on darunavir/ritonavir or atazanavir/ritonavir based ART to 120mg FDV QD. Importantly the study also allowed the inclusion of patients with liver cirrhosis allowing to gain some efficacy and safety data in this more difficult to treat patient population. Overall 304 (239 treatment naïve and 69 relapsers) patients were included making this the largest coinfection DAA study so far. Importantly, 17% of patients included had F4 fibrosis, 78% GT1a and baseline HCV RNA was ≥800 000 IU/mL in 80% of patients. Final SVR rates in HIV/HCV co-infected patients are depicted in figure 10.
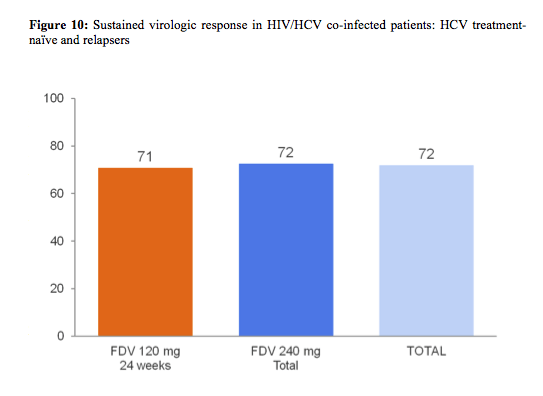
FDV was highly efficacious and well tolerated in difficult-to-treat patients co-infected with HIV and HCV GT-1 and resulted in a total SVR12 rate of 72%. A high proportion of patients (80%) achieved 'early treatment success' (ETS) and 86% of these patients achieved SVR12. Interestingly, high SVR12 rates were achieved regardless of HCV subtype, presence of compensated cirrhosis or FDV doses and durations studied. The safety profile of FDV in HIV and HCV GT-1 co-infected patients was similar to that observed in HCV GT-1 mono-infected patients. Adverse events leading to treatment discontinuation occurred in 7% of patients, discontinuation because of faldaprevir associated adverse events only in 1% of patients.
More interesting results from the PK-sub studies were presented in the themed discussion session "Clinical Pharmacology in Patient care" as well as in the poster session (11, 12). In studies in healthy volunteers, FDV increased raltegravir (RAL) exposure by about 2.5-fold and had no substantial effect on the pharmacokinetics (PK) of darunavir/ritonavir (DRV/r), efavirenz (EFV), or tenofovir darunavir/ritonavir (DRV/r), efavirenz (EFV), or tenofovir (TDF). In these sub studies PK interactions between FDV and the antiretrovirals (ARV) used in the STARTVerso4 study (the phase 3 study described above) were analyzed. Of 308 treated patients, 297 were receiving ARV therapy (ATV/r [n=12], DRV/r [n=55], EFV [n=84], RAL [n=143], or other ARV [n=3]). There was no apparent effect of FDV on EFV C12. DRV gMean trough level was lower with FDV than without FDV whereas RAL gMean trough level was similar with or without FDV in the 120 mg group but lower with FDV than without FDV in the 240 mg group. TDF gMean trough levels were lower with than without FDV in the 120 mg group and in the240 mg group in the absence of EFV. In the presence of EFV, TDF trough levels in the FDV 240 mg group were comparable with or without FDV. A substudy with intensive PK sampling showed no effect of FDV on ATV PK parameters. In summary, in this pk analysis from STARTVerso4, the pk results were consistent with observations from studies in healthy volunteers; FDV had no clinically relevant effect on the studied ARVs in patients co-infected with HIV and HCV.
The next study with a new HCV second wave protease inhibitor was the C212 study, which studied simeprevir in combination with PEG-IFN and ribavirin (5). The study design is shown below (figure 11):
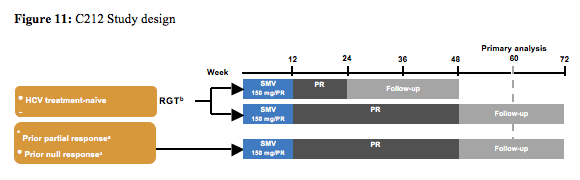
Primary endpoint of the study was SVR12, safety, and tolerability. Secondary endpoints: virologic response at other time points, on-treatment failure, and relapse rates. Patients were either receiving HAART (abacavir [ABC], tenofovir [TDF], lamivudine [3TC], emtricitabine [FTC], rilpivirine (RPV), raltegravir [RAL], maraviroc [MVC], or enfuvirtide [ENF]) or not receiving HAART. For patients on ART vs those not on ART: Baseline CD4+ count: 561 vs 677 cells/mm3; Baseline HIV RNA: <50 copies/mL 88% vs 15%; <200 copies/mL 98% vs 23%. 12% of patients at baseline had F3 fibrosis and 9% F4. The final efficacy results are shown in figure 12.
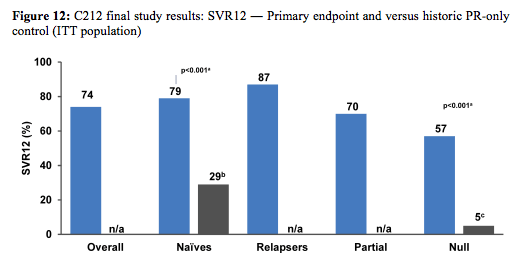
Simeprevir 150 mg QD + pegIFN/RBV led to high virologic response rates in co-infected patients, in particular treatment naïve with 79% SVR12 and 87% SVR12 in prior relapsers. In previous null responders however, overall lower cure rates of 57% were observed emphasizing that more difficult to treat patients may require more potent strategies such as 2 DAA combinations. Patients without cirrhosis who met RGT criteria: 89% (54/61) of patients met RGT criteria for 24 weeks of treatment (41/48 treatment-naïve patients; 13/13 prior relapsers). Interestingly the presence of the Q80K polymorphism at baseline had almost no impact on SVR rates in GT1a patients. The safety profile was similar to that observed in HCV mono-infected patients. The frequency of grade 3 / 4 AEs and SAEs was similar in patients with/without ART. Overall only 4 patients discontinued simeprevir due to adverse events.
In conclusion the two second wave HCV protease inhibitor studies with simeprevir and faldaprevir respectively, demonstrate that new HCV protease inhibitors will become or are already available which offer once daily dosing, improved tolerability profile and most importantly shortened treatment duration for the majority of patients who achieve RVR. Also the drug interaction profile is different and at least with faldaprevir the combination with boosted darunavir is possible. Nevertheless, treatment duration with these compounds in combination with PEG-IFN/RBV is longer than with sofosbuvir + PEG-IFN/RBV making it unlikely that these compounds will have a role in IFN based HCV treatment regimens. They may however, be interesting as DAA combination partners in 2-3 IFN-free DAA combinations and also play a role depending on price and overall DAA availability in a given country, respectively.
New data on interferon-free HCV treatment approaches in HIV/HCV coinfection
Clearly, the ultimate goal in treatment of hepatitis C in HCV monoinfected just as well as in HIV coinfected subjects is to develop interferon-free DAA regimens which allow cure in the majority of patients and because of less toxicity and shorter treatment durations will revolutionize HCV management within the next years.
Clearly with regard to coinfection studies the final results from the first interferon-free trial in coinfection were among the most exciting study results presented at CROI (6). Interim analyses of the PHOTON 1 study had already previously been presented at last year AASLD (13). The PHOTON 1 study evaluated the safety and efficacy of the oral HCV NS5B inhibitor sofosbuvir (SOF) with ribavirin (RBV) in HIV -seropositive patients coinfected with HCV genotypes 1, 2 or 3. Patients included into the study received SOF 400 mg QD and RBV 1000-1200mg/day; GT 1 treatment-naïve (TN) patients received 24 weeks and GT 2/3 treatment-naïve patients received 12 weeks of treatment. Treatment experienced GT 2/3 patients received 24 weeks of therapy. The study design is shown below in figure 13.
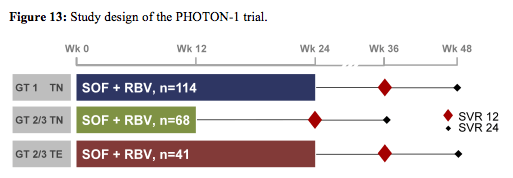
Overall, 4% of the treatment -naïve GT1 patients (n=114), 4% of the TN GT2 patients (n=26) and 14% of the TN GT 3 patients (n=42), respectively had cirrhosis at baseline. 24% of the treatment-experienced GT 2/3 patients had cirrhosis at baseline making this clearly the most difficult to treat patient group. 93-98% of patients were on ART and had stable HIV disease. The final SVR rates are shown below in figure 14. Amazingly in this first IFN-free study in HIV/HCV coinfection SVR12 was achieved in 76% (87/114) of TN GT1, 88% (23/26) of TN GT2 and 67% (28/42) in TN GT3, respectively (see figure 14). Extending treatment duration to 24 weeks in treatment experienced GT 2/3 substantially increased response rates in GT3 patients to 94%.
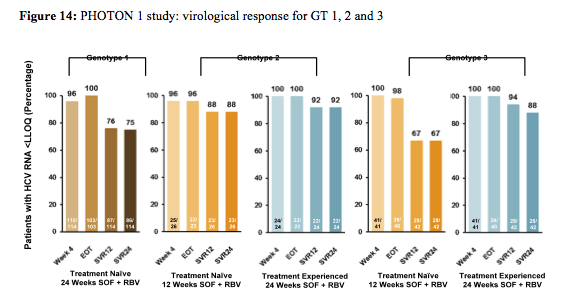
Among the 27 TN GT 1 patients without SVR12, viral relapse was confirmed in 25 patients. In contrast no relapse occurred in the GT 2 patients but in 12 TN GT 3 patients without SVR12. Therefore it appears that treatment duration of 12 weeks is sufficient for GT2 patients whereas for GT3 potentially longer treatment durations are required to prevent relapse. Indeed the prolonged treatment in the treatment experienced GT3 patients decreased relapse rate substantially with only one documented relapse after stopping therapy. For treatment-naïve GT1 the majority of patients respond well to an interferon-free regimen of sofosbuvir and ribavirin but the high relapse rate suggests that there exists a subset of patients which will require DAA combination therapy. Overall, these results demonstrate that interferon-free treatment strategies are a very close reality for the majority of patients. As response rates are virtually superimposable to findings in monoinfected patients these findings also underline that probably HIV patients no longer should be treated as an independent patient population (other than for their potential for drug-drug interactions between HCV drugs and antiretroviral therapy) but on average can be treated like HCV mono-infected subjects.
The safety findings of the study are summarized in table 1:

Overall a majority of patients reported some adverse events, although no clear difference emerged between treatment lengths. For adverse events reported in >/= 10% there was no clear difference between the 24 and 12 week groups. Grade 3-4 adverse events occurred in 12 and 10% of patients and serious adverse events in 3 and 4% of patients. Treatment discontinuation due to adverse events was uncommon (3 and 4%) and these included weight loss, insomnia/agitation, pneumonia, suicide attempt, foreign body sensation in throat, increased anxiety, and dyspnea. There was one death in the 12 week treatment arm, which was a suicide 9 days after completing treatment.
In summary, the interferon-free regimen of SOF + RBV resulted in high SVR12 and SVR24 rates in HIV-infected patients with HCV genotype 1, 2 and 3 co-infection. SVR12 rates were similar to those observed in patients with HCV monoinfection. No resistance was observed in virologic failures. SOF was well tolerated, with a low rate of treatment discontinuations due to adverse events. Clearly for GT2 patients this is already the ideal regimen promising cure for almost all patients after 12 weeks of therapy. For GT 3 patients extending duration to 24 weeks clearly is the preferred schedule with much lower relapse rates than observed with 12 weeks. An alternative to the 24 weeks of all oral therapy could be 12 weeks of sofosbuvir + PEG-IFN/RBV in GT3 patients with no contraindication or previously documented intolerance to IFN.
The second IFN free study in coinfection was presented as a late-breaker poster (7). This particular study compared the on-treatment response of HIV/HCV coinfected patients treated in the C-WORTHY study Part B to the previously reported monoinfected cohort of patients in Part A of the C-WORTHY study (see also study design figure 15). Part A of the C-WORTHY evaluated a 12 week regimen of MK-5172 (an NS3/4A protease inhibitor)/MK-8742 (an NS5A inhibitor) ± RBV among 65 GT1 HCV monoinfected, treatment naïve, non-cirrhotic patients. In this group a SVR was achieved in 96-100%. Only one virologic failure was documented as a relapse at follow-up week 4. Overall, 3 subjects discontinued prior to Week 5 for non-AE related reasons. Part B of C-WORTHY evaluated a 12 week regimen of MK-5172/MK-8742 ± RBV among 403 GT1-infected patients, including 59 HIV/HCV GT1 coinfected, treatment naïve, non-cirrhotic patients.
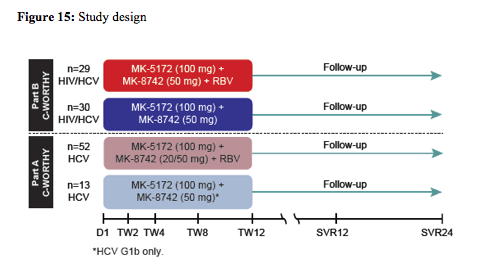
With regard to ART patients had to be stable on raltegravir + two NRTIs for at least 8 weeks prior to enrollment to be included into the study. ART dose modification were not permitted during 8 weeks prior to enrollment unless dose modification due to treatment failure. CD4 cell count was required to be >300 cells/mm3 and HIV RNA undetectable for 24 weeks. The on-treatment response rate for HIV/HCV coinfected versus HCV monoinfected subjects is depicted in figure 16:
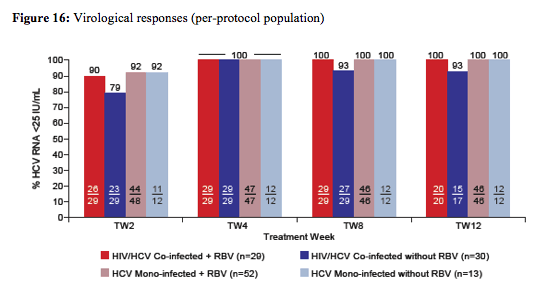
The per-protocol population was defined as the primary study population efficacy evaluation. Overall 11 monoinfected and one coinfected patient were excluded from the per protocol population (11 Monoinfected: 7 Pts excluded (4 Pts received wrong RBV dose; all achieved SVR12); 3 Pts discontinued early (Day 3, 22, 35)-they will be included in the analysis up to time of discontinuation; 1 Coinfected: 1 Pt excluded (lost to follow-up after TW4). Overall safety was good and no patient discontinued the study drugs due to laboratory abnormalities or adverse events.
In conclusion, among HCV genotype 1 infected patients with and without HIV, the combination of MK-5172 and MK-8742 ± RBV was associated with robust HCV suppression on treatment. Final efficacy will be determined by viral responses during follow-up.
New data on interferon-free HCV treatment approaches in HCV monoinfection
At this year's CROI for the first time data from the IFN free combination of simeprevir and daclatasvir was presented (14). Under consideration of the fact that simeprevir is already licensed in the US and both simeprevir and daclatasvir will become available in Europe this could be an interesting IFN free DAA combination. The study design of the so-called LEAGUE study is depicted in figure 17:
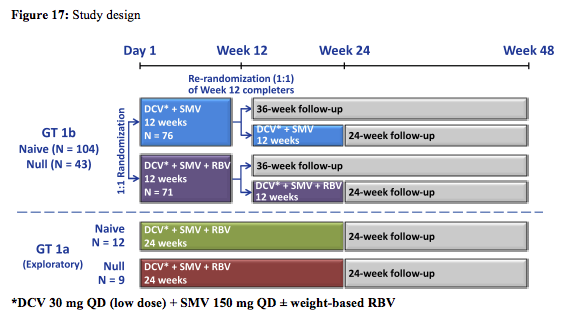
It is important to highlight that DCV dose selection was based on PK data in healthy volunteers showing a 2-fold increase in DCV (60 mg) exposure when dosed in combination with SMV (150 mg). Therefore, the lower 30mg dose was chosen. In this study, DCV exposure due to co-administration with SMV was less than anticipated and may therefore have impacted the results.
The study required the inclusion of men and women aged ≥ 18 years infected with HCV GT 1a or 1b. Treatment-naïve was defined as no prior HCV treatment, null responder as never achieved ≥ 2 log10 decline in HCV RNA after ≥12 weeks of peginterferon/RBV treatment. Patients with advanced fibrosis/cirrhosis were allowed to be included but capped at 35% with ≥ 20% F4. The primary efficacy endpoint was proportion of patients with sustained virologic response at posttreatment Week 12 (SVR12). The efficacy outcomes for GT 1b are shown in figure 18:
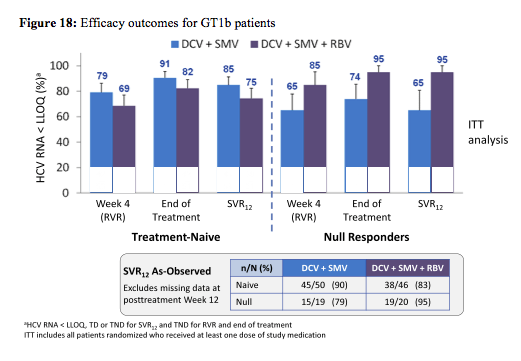
The all-oral combination of DCV 30-mg QD (low dose) + SMV 150-mg QD ± RBV achieved SVR12 rates of 75-85% in treatment-naïve patients and 65-95% in prior null responders with GT 1b infection. Treatment duration did not have an impact on probability of SVR suggesting 12 weeks to be sufficient for this combination. In null responders the impression arises that addition of ribavirin increased SVR rate. The distribution of virological failures is shown in figure 19:
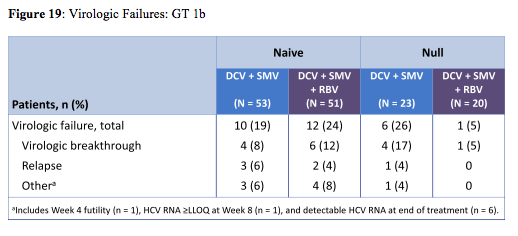
The considerable rate of virological failures throughout most arms suggests that this combination (potentially because of the low dose) may not be the ideal solution for all patients.
The exploratory arm in GT1a patients revealed a 67% SVR12 rate in treatment-naïve patients including 2/2 cirrhotic and 6/10 non-cirrhotic patients. Virologic breakthrough was experienced by 4/12 (33%) patients. GT 1a null responders were offered the addition of peg-interferon to DCV + SMV + RBV after 5 patients had experienced virologic breakthrough. Overall, a total of 7 patients experienced virologic breakthrough, all patients (n=9) in this cohort were analyzed as non-SVR. So clearly for GT1a non-responders this is not a recommendable combination. The safety in all arms was very good with few discontinuations because of adverse events (2-3%). Further exploration of this combination with the higher dose of simeprevir appears necessary before final conclusions on the efficacy of this regimen can be drawn.
The all-oral triple combination of daclatasvir (DCV; NS5A inhibitor), asunaprevir (ASV; NS3 inhibitor), and BMS-791325 ('325; nonnucleoside NS5B inhibitor) achieved sustained virologic response (SVR) rates >90% in pilot cohorts of non-cirrhotic patients with chronic HCV genotype (GT)1 infection. At this year's CROI the study expansion (AI443-014) evaluated this regimen in larger cohorts that included cirrhotic patients, with all components
dosed BID to support coformulation development (15). Overall, 166 treatment-naïve, HCV GT1-infected patients were randomly assigned (1:1) to receive a twice-daily regimen of DCV 30mg, ASV 200mg, and '325 75mg (n=80) or 150mg (n=86) for 12 weeks. Randomization was stratified by GT1 subtype and presence of cirrhosis. The primary endpoint was HCV RNA < LLOQ (25 IU/mL) at 12 weeks post treatment (SVR12). The study design is depicted below in figure 20:
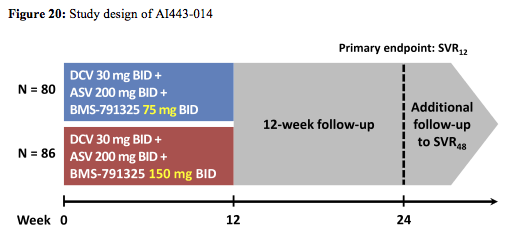
Baseline characteristics were comparable across treatment groups; overall, patients were 56% male, 83% white, 82% GT1a, 67% IL28B non-CC genotype, and 9% cirrhotic. The virological treatment outcome is depicted below in figure 21:
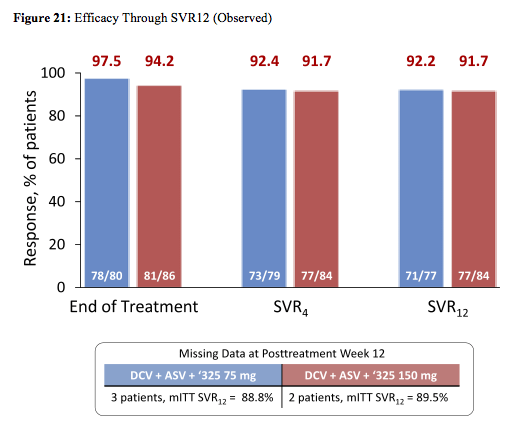
High SVR12 rates were observed regardless of 325 doses. Response rates by chirrosis stage or IL28b are shown in figure 22:
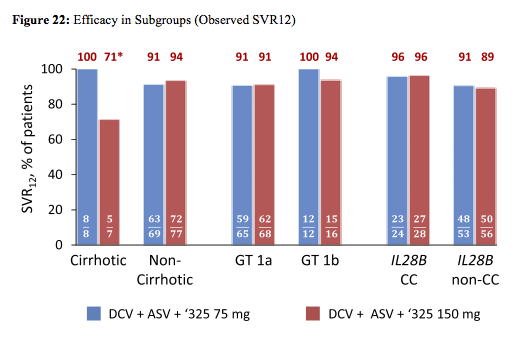
*2 cirrhotic patients added pegIFNα/RBV: 1 viral breakthrough; 1 met a protocol stopping rule for '325.(bilirubin increase).
Within the entire study population only 5/166 patients experienced on-treatment virologic breakthrough and, to date, 6/166 relapsed posttreatment; all of these patients had GT1a infection. Of 11 failures, 5 patients had emergent resistance to DCV and ASV; 6 patients had 3-drug resistance. Presence at baseline of GT 1a resistance-associated polymorphisms in NS3 (n=66), NS5A (n=29), or NS5B (n=15) sequences was not clearly predictive of failure. Signature resistance-associated polymorphisms detected at failure included: NS3: V36M, T54S, R155K, NS5A: M28T, Q30E/H/R, L31M, Y93H/N and NS5B: P495L/S. In each group, 1 patient discontinued due to an AE (unrelated esophageal neoplasm, throat tightness); there were no deaths and no treatment-related serious AEs or grade 3/4 AEs. The most frequent AEs in both groups were headache, diarrhea, fatigue, and nausea. The only grade 3/4 liver-related lab abnormalities were single events of elevated AST (grade 3) and total bilirubin (grade 3) in different patients.
In summary, 12 weeks of all-oral treatment with DCV + ASV + BMS-791325 achieved an overall SVR12 rate of 92% in a predominantly GT 1a population, with comparable response rates in cirrhotic and non-cirrhotic patients and low rates of virologic failure. The regimen was generally well tolerated, with similar safety profiles for both '325 dose groups. Further evaluation of this triple regimen is ongoing in broader patient populations, including GT4 infection and prior GT1 null responders.
Another very important IFN-free DAA study which was the presented at CROI was the PEARL III study (16). The PEARL III study is a multinational phase 3 trial of co-formulated ABT-450/r/ABT-267 and ABT-333 (3D regimen), with and without RBV, in non-cirrhotic treatment-naïve adults with GT1b HCV infection. ABT-450 is an HCV protease inhibitor (dosed with ritonavir 100mg, ABT-450/r) identified by AbbVie and Enanta. ABT-267 is an NS5A inhibitor, and ABT-333 is a nonnucleoside polymerase inhibitor. The study design is shown below in figure 23:
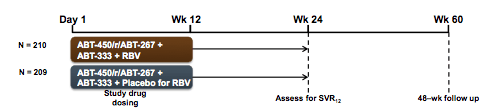
Subjects were randomized (1:1) to 12 weeks of treatment with ABT-450/r/ABT-267 (150mg/100mg/25mg QD) and ABT-333 (250mg BID), with weight-based RBV (1000mg or 1200mg daily divided BID, Arm A) or placebo for RBV (Arm B). 419 subjects received the 3 drug regimen, baseline characteristics were well balanced between the two arms. Only 21% in both arms had an IL28b CC genotype. 9.6-10.5% of patients had F3 fibrosis at baseline. Primary endpoint was non-inferiority of SVR12 rate for each regimen to the historical rate achieved in a similar population with telaprevir + pegIFN/RBV SVR12 rates (intent-to-treat). Virological outcome of the trial is shown in figure 24:
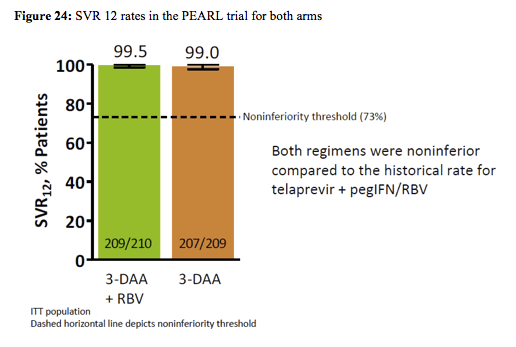
99.5% (Arm A) and 99.0% (Arm B.) achieved HCV cure. There was no on-treatment virologic failure or post-treatment relapse among subjects receiving the 3D regimen without RBV. Only 1 patient on the 3 DAA + RBV combination developed virologic rebound at week 10 with emergence of Y93H variant subjects in NS5A. 19 subjects in Arm A and 0 in Arm B (P<0.001) had hemoglobin decrease to <10g/dL. The most common adverse events in Arms A and B were headache (24.3% vs. 23.4%, P=NS) and fatigue (21.4% vs. 23.0%, P=NS.) No subjects discontinued due to adverse events.
In conclusion this large phase 3 trial demonstrated that the 3 drug combination of ABT-450/r/ABT-267 and ABT-333 was highly efficacious and safe with or without RBV for the treatment of HCV GT1b infected non-cirrhotic treatment-naïve adults. There is no need for RBV in this population.
The last but clearly most exciting study to report back from the oral hepatitis session from CROI was the NIH SYNERGY study on shorter HCV treatment durations (17). In this pilot trial sixty HCV mono-infected, treatment naïve, GT-1, patients were consecutively enrolled into 3 arms of a phase 2 prospective cohort study and received: Arm A - sofosbuvir with ledipasvir (400mg/90mg respectively once daily in a fixed dose combination (FDC)) for 12 weeks, Arm B - FDC + GS-9669 (500mg/day), a non-nucleoside NS5B inhibitor for 6 weeks, or Arm C - FDC + GS-9451 (80mg/day), an HCV protease inhibitor for 6 weeks. The study design is also shown below in figure 25:
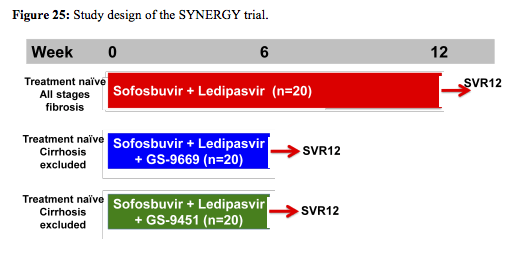
Serial measurements of safety parameters, virologic (HCV RNA by Roche Taqman PCR and Abbott assay; deep sequencing of baseline mutations) and host correlates (intrahepatic and peripheral by flow cytometry) were performed. Of note the study population represented a more challenging to treat patient group with potential adherence challenges. Patients enrolled were predominantly African American (88%), male (72%), infected with GT-1a (70%), had a high HCV VL (>800k) (70%) with an IL28B non-CC haplotype (82%). Baseline demographics were similar between patients across arms and 35%, 25% and 25% of subjects on Arm A, B and C, respectively, had stage ≥3 liver fibrosis. No patients with cirrhosis were included in Arm B or C. The virological outcome of the trial is shown below in figure 26:
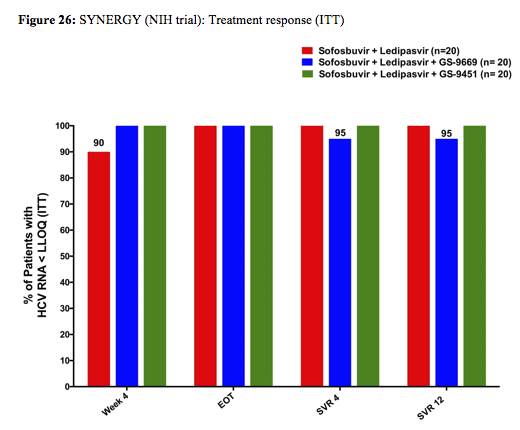
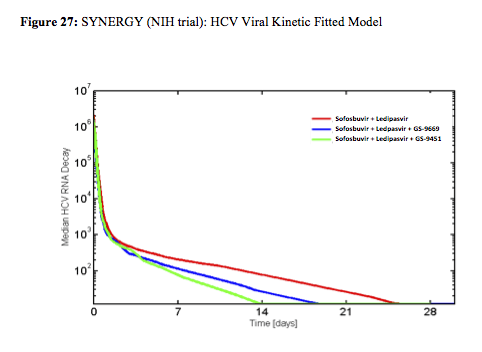
The high SVR12 rates clearly document that in 3DAA therapy (without IFN or ribavirin) shortened treatment durations of 6 week are possible. Also the 100% response rate at week 6 in arm C suggests that even shorter treatment should be feasible at least in a subset of patients. Only one patient in arm B relapsed. Most interestingly the authors presented data from their viral kinetic sub-study suggesting that patients in the 3 DAA arms reach undetectability faster than the patients in the 2DAA arm which may explain why they can be treated shorter (see figure 27). The real value of this study is that particularly for patient populations with adherence issues there may be hope for successful treatment outcome after very short cycles of therapy. Indeed in a further poster the same group demonstrated that increased pill burden and longer treatment durations (particularly between week 8 and 12) in a urban clinic cohort was accompanied by a decrease in adherence (18).
The combination was well tolerated with no grade 4 adverse events or drug discontinuations.
The authors concluded that in this inner city patient population addition of a third antiviral agent allowed successful eradication of HCV in 6 weeks in a difficult to treat patient population. This study thereby represents a new paradigm of combination therapy to reduce HCV treatment duration, which may be vital in the treatment and eradication of HCV globally.
Any other interesting news?
There were some studies looking at the outcome of acute HCV in HIV coinfected msm who did not receive or not respond to treatment during acute infection (19). Overall, 41 HIV-infected patients from 3 European countries with diagnosed acute HCV infection since 2003 with at least 12 months of follow-up and persistent HCV viremia were repeatedly evaluated for liver fibrosis by means of transient elastography. Interstingly, there was no significant change in median liver stiffness over a maximum follow-up of 8 years. There was also no significant correlation between liver stiffness and reason for HCV persistence (chronic course vs. unsuccessful treatment), follow-up time, BMI, alcohol or drug abuse, diabetes, lipodystrophy, cART duration, or exposure to d-drugs.
In conclusion, an episode of acute hepatitis C in HIV-positive MSM does not appear to lead towards early advanced liver fibrosis. This finding is particularly reassuring for clinicians and patients in whom HCV persists due to a chronic course or unsuccessful early treatment
Summary
· Studies on the efficacy and safety of the HCV protease inhibitors boceprevir or telaprevir in HCV genotype 1 HIV co-infected individuals who have previously shown a non-response under dual HCV therapy with PEG-IFN/RBV have clearly demonstrated surprisingly high SVR rates. These data underline that even in more difficult to treat patients with previous non-response to dual therapy and more advanced fibrosis stages HCV cure is achievable in a considerable amount of patients and therefore needs to be considered in patients who may develop HCC or decompensate prior to the availability of interferon-free HCV therapies in a respective country where cost issues have delayed the access to more recent DAAs. It needs to to be highlighted though that these first DAA based therapies come with a high pill burden and increased haematological toxicities. Clearly getting access to better tolerated and easier to apply HCV DAAs should remain the most important priority.
· HCV treatment including telaprevir or boceprevir in more advanced patient populations (higher fibrosis stages) may be associated with a higher rate of haematological toxicities and warrants careful monitoring and aggressive anemia management (ribavirin dose reduction or EPO use)
· Telaprevir administration can decrease GFR and thereby increase ribavirin levels potentially enhancing anemia.
· The two second wave HCV protease inhibitor studies in HIV/HCV coinfected individuals with simeprevir and feldaprevir, respectively have demonstrated high SVR rates in 72-74% of patients. The majority of patients achieved RVR and were able to be randomized to shorter treatment durations of 24 weeks.
· Results from the first interferon-free trial in HIV/HCV coinfection demonstrate that the majority of GT1 may be curable with the all oral sofosbuvir plus ribavirin combination for 24 weeks. Clearly 12 weeks of this combination are sufficient for treatment in GT 2 patients. For GT 3 patients extending therapy to 24 weeks promises cure for most patients. There will be a need for DAA combination therapy in around a quarter of GT 1 patients reflecting the relatively high relapse rate after treatment discontinuation.
· The combination of MK-5172 and MK-8742 ± RBV was associated with robust HCV suppression on treatment which was comparable between HIV/HCV coinfected and HCV mono-infected subjects. Final efficacy will be determined by viral responses during follow-up.
· The all-oral combination of DCV + SMV[low dose of DCV], with or without RBV, was generally well tolerated and achieved SVR12 in 75-85% of treatment-naïve patients and 65-95% of prior null responders with GT1b infection after 12 or 24 weeks of treatment. .In GT1a patients this combination only achieved a 67% SVR rate in the treatment naïve patients whereas all previous null respondeers did not respond suggesting this may not be the best option in this particular patient group.
· 12 weeks of all-oral treatment with DCV + ASV + BMS-791325 achieved an overall SVR12 rate of 92% in a predominantly GT 1a population, with comparable response rates in cirrhotic and non-cirrhotic patients and low rates of virologic failure. The regimen was generally well tolerated, with similar safety profiles for both '325 dose groups.
· The 3 drug combination of ABT-450/r/ABT-267 and ABT-333 was highly efficacious and safe with or without RBV for the treatment of HCV GT1b infected non-cirrhotic treatment-naïve adults. There is no need for RBV in this population.
· In the SYNERGY trial the addition of a third antiviral agent allowed successful eradication of HCV in 6 weeks in a difficult to treat patient population. This study thereby represents a new paradigm of combination therapy to reduce HCV treatment duration, which may be vital in the treatment and eradication of HCV globally.
References
1. Cotte L, Braun J, Vincent C, Sogni P, et al.; Telaprevir in Treatment-Experienced HIV-HCV G1 Coinfected Patients (ANRS HC26 TelapreVIH). 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 668
2. Poizot-Martin I, Bellissant E, Colson P, et al., and the ANRS-HC27 BOCEPREVIH Study Group. ANRS-HC27 Boceprevir for Previously Treated HCV-HIV Coinfected Patients: The ANRS-HC27 BocepreVIH Trial. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2013; abstract 659LB
3. Neukam K, Munteanu D, Rivero A et al.; Boceprevir/Telaprevir-Based Therapy in HIV-Infection: Interim Analysis of a Multicenter Cohort. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 660
4. Dieterich D, Tural C, Nelson M, et al.; Branch A. Faldaprevir Plus Pegylated Interferon Alfa-2a/Ribavirin in HIV/HCV Coinfection: STARTVerso4. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 23
5. Dieterich D, Rockstroh J, Orkin C, et al.; Simeprevir (TMC435) Plus PegIFN/Ribavirin in HCV Genotype-1/HIV-1 Coinfection (Study C212). 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 24
6. Naggie S, Sulkowski M, Lelazeri J, et al.; Sofosbuvir Plus Ribavirin for HCV Genotype 1-3 Infection in HIV Coinfected Patients (PHOTON-1). 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 26
7. Sulkowski M, Mallolas J, Bourliere M, et al.; On-Treatment Viral Response To MK-5172/MK-8742 ± RBV for 12 Weeks in HCV/HIV-Coinfected Patients. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 654LB
8. Cotte L, Barrail-Tran A, Vincent C, et al.; Telaprevir Increases Ribavirin Toxicity Through eGFR Decrease in HIV-HCV Coinfected Patients21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 664LB
9. Haubitz S, Schaerer V, Ambrosioni J, et al.; Protease Inhibitors To Treat Hepatitis C in the Swiss HIV Cohort Study: High Efficacy But Low Uptake. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 658
10. Gori A, Doroana M, Chernova O, et al.; Telaprevir Treatment of HIV/HCV G1 Patients with Severe Fibrosis: Efficacy Results to Week 16. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 663
11. Rockstroh J, Valantin MA, Mallolas JM, et al.; Pharmacokinetics of Faldaprevir and Antiretrovirals in Patients With HIV/HCV Coinfection 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 497
12. Nelson M, Arasteh K, Jain MK et al.; Effect of Faldaprevir On Atazanavir Pharmacokinetics in Patients With HIV/HCV Coinfection
13. Sulkowski M et al., All-Oral Therapy With Sofosbuvir Plus Ribavirin For the Treatment of HCV Genotype 1, 2, and 3 Infection in Patients Co-infected With HIV (PHOTON-1). 64th Annual Meeting of the American Association for the Study of Liver diseases, November 1-5, 2013, Washington ,USA; abstract 212
14. Zeuzem S, Hezode C, Bronowicki JPP et al.; Daclatasvir in Combination with Simeprevir ± Ribavirin for Hepatitis C Virus Genotype 1 Infection. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 28LB
15. Everson GT, Thuluvath PJ, Lawitz E, et al.; All-Oral Combination of Daclatasvir, Asunaprevir, and BMS-791325 for HCV Genotype 1 Infection. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 25
16. Ferenci P et al.; PEARL III: SVR ≥99% After 12 Wks of ABT-450/r/267 + ABT-333 ± RBV in Treatment Naïve HCV GT1b Infection. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 29LB
17. Kohli A, Sims Z, Nelson A et al.; Combination Oral, Hepatitis C Antiviral Therapy for 6 or 12 Weeks: Final Results of the SYNERGY Trial. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 27LB
18. Petersen T, Gordon LA, Townsend K, et al.; Pill Burden & Treatment Length Reduce Adherence To IFN-Free Hepatitis C Therapy in an Urban Cohort. 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 667
19. Boesecke C, Ingiliz P, Mandorfer M et al.; and the NEAT Study Group. Is There Long-Term Evidence of Advanced Liver Fibrosis After Acute Hepatitis C in HIV Coinfection? 21st Conference on Retroviruses and Opportunistic Infections, March 3-6, 2014; abstract 664
|
| |
|
|
|
|
|