| |
Virological response after 6 week triple-drug regimens for hepatitis C: a proof-of-concept phase 2A cohort study
|
| |
| |
Dowload the PDF here
"In our study, patients with chronic HCV genotype 1 infection without cirrhosis were successfully treated with a 6 week course of three oral direct-acting antiviral drugs.....Participants were mainly black (53, 88%), men (43, 72%), had IL28 non-CC genotype (48, 80%), were infected with HCV genotype 1a (42, 70%), and had high baseline concentrations of plasma HCV RNA (>800 000 IU/mL; 42, 70%). 15 (25%) patients had stage 3 liver disease. Of patients treated with sofosbuvir and ledipasvir, three (15%) had stage 4 disease (table 1), no patients had stage 4 disease in the other groups."
"we estimated the effect of overall treatment on HCV clearance by taking into account the effectiveness of the regimen in blocking HCV production (δ1 and δ2) and intracellular decay (κ). Median overall treatment effect for all three regimens did not differ significantly (p>0·05; 99·977% for sofusbuvir and ledipasvir, 99·954% for sofosbuvir, ledipasvir, and GS-9669, and 99·984% for sofosbuvir, ledipasvir, and GS-9451; appendix). Nevertheless, more patients in the group given sofosbuvir, ledipasvir, and GS-9451 had a maximum threshold effect of 99·95% or higher in clearance of plasma HCV than did those in the group given the other regimens (p=0·09)."
(new Study 3 DAAs) Sovaldi \ ledipasvir+vedroprevir Study in treatment-experienced gt1 cirrhotics - 8 weeks - (10/08/14)
(this study was presented at CROI/2014) Combination Oral, Hepatitis C Antiviral Therapy for 6 or 12 Weeks: Results of the SYNERGY Trial.....http://www.natap.org/2014/CROI/croi_04.htm
Safety, Tolerability, Pharmacokinetics, and Antiviral Activity of GS-9857 (pan-genotypic protease) in Subjects With Chronic Hepatitis C Virus Infection......https://clinicaltrials.gov/ct2/show/NCT02185794
--------------------------
Virological response after 6 week triple-drug regimens for hepatitis C: a proof-of-concept phase 2A cohort study
The Lancet January 13, 2015
Anita Kohli, Anuoluwapo Osinusi, Zayani Sims, Amy Nelson, Eric G Meissner, Lisa L Barrett, Dimitra Bon, Miriam M Marti, Rachel Silk,
Colleen Kotb, Chloe Gross, Tim A Jolley, Sreetha Sidharthan, Tess Petersen, Kerry Townsend, D'Andrea Egerson, Rama Kapoor, Emily Spurlin,
Michael Sneller, Michael Proschan, Eva Herrmann, Richard Kwan, Gebeyehu Teferi, Rohit Talwani, Gabbie Diaz, David E Kleiner, Brad J Wood,
Jose Chavez, Stephen Abbott, William T Symonds, G Mani Subramanian, Phillip S Pang, John McHutchison, Michael A Polis, Anthony S Fauci,
Henry Masur, Shyam Kottilil
Summary
Background
Direct-acting antiviral drugs have a high cure rate and favourable tolerability for patients with hepatitis C virus (HCV). Shorter courses could improve affordability and adherence. Sofosbuvir and ledipasvir with ribavirin have high efficacy when taken for 8 weeks but not for 6 weeks. We assessed whether the addition of a third direct-acting antiviral drug to sofosbuvir and ledipasvir would allow a shorter treatment duration.
Methods
In this single-centre, open-label, phase 2A trial, we sequentially enrolled treatment-naive patients with HCV genotype 1 infection into three treatment groups: 12 weeks of sofosbuvir and ledipasvir; 6 weeks of sofosbuvir, ledipasvir, and GS-9669; or 6 weeks of sofosbuvir, ledipasvir, and GS-9451. Patients and investigators were not masked to treatment assignment. The primary endpoint was the propotion of patients with sustained viral response at 12 weeks after treatment completion (SVR12), assessed by serum HCV RNA concentrations lower than 43 IU/mL (the lower limit of quantification). We did an intention-to-treat analysis for the primary endpoint and adverse events. This study is registered with ClinicalTrials.gov, number NCT01805882.
Findings
Between Jan 11, 2013, and Dec 17, 2013, we enrolled 60 patients, and sequentially assigned them into three groups of 20. We noted an SVR12 in all 20 patients (100%, 95% CI 83-100) allocated to sofosbuvir and ledipasvir for 12 weeks; in 19 (95%, 75-100) of the 20 patients allocated to sofosbuvir, ledipasvir, and GS-9669 for 6 weeks (one patient relapsed 2 weeks after completion of treatment); and in 19 (95%, 75-100%) of the 20 patients allocated to sofosbuvir, ledipasvir, and GS-9451 for 6 weeks (one patient was lost to follow-up after reaching sustained viral response at 4 weeks). Most adverse events were mild and no patients discontinued treatment. Two serious adverse events occurred (pain after a post-treatment liver biopsy and vertigo), both unrelated to study drugs.
Interpretation
In this small proof-of-concept study, two different three-drug regimens that were given for 6 weeks resulted in high cure rates for HCV infection with excellent tolerability. Addition of a third potent direct-acting antiviral drug can reduce the duration of treatment required to achieve sustained viral response in patients with chronic HCV genotype 1 infection without cirrhosis.
Funding
National Institute of Allergy and Infectious Diseases (NIAID), National Cancer Institute and Clinical Center Intramural Program, German Research Foundation, National Institutes of Health, Gilead Sciences.
Introduction
About 185 million people worldwide are infected with hepatitis C virus (HCV).1 Up to 20% of these patients develop cirrhosis and a quarter of those will progress to end-stage liver disease or hepatocellular carcinoma.2 Until late 2013, the treatment for hepatitis C genotype 1 was combination therapy with pegylated interferon, ribavirin, and, more recently, a direct-acting antiviral drug, for up to 1 year.3, 4, 5 These regimens are difficult to tolerate because of adverse effects associated with each constituent of the triple-drug regimens, with efficacy of 56-88%.4, 5 In 2013, the US FDA licensed two new direct-acting antiviral drugs, sofosbuvir and simeprevir, for the treatment of HCV infections as part of combination regimens. In addition, 91-100% of patients given sofosbuvir and the antiviral drug ledipasvir as a one pill per day regimen for 12 weeks had sustained viral response (SVR).6, 7 Regimens with a short duration, low pill burdens, and few adverse effects could improve patient adherence.
An attempt to reduce the duration of therapy to 6 weeks through the addition of ribavirin to sofosbuvir and ledipasvir resulted in many patients relapsing after treatment.8 We postulated that addition of a third direct-acting antiviral drug instead of ribavirin to sofosbuvir and ledipasvir could allow for shorter, more efficacious, treatment. We did a three-group clinical trial in a predominantly male black population, aiming to compare SVR prevalence for patients given sofosbuvir and ledipasvir for 12 weeks, sofosbuvir and ledipasvir plus GS-9669 (a non-nucleoside NS5B thumb site 3 inhibitor of HCV polymerase8, 9) for 6 weeks, or sofosbuvir and ledipasvir plus GS-9451 (an inhibitor of the HCV NS3/4A protease10) for 6 weeks.
Methods
Participants
We enrolled patients at the Clinical Research Center of the National Institutes of Health (NIH), Bethesda, MD, USA. We report enrolment and follow-up data from Jan 11, 2013, to Dec 17, 2013. Eligible participants were aged 18 years or older, infected with chronic HCV genotype 1 (serum HCV RNA concentration �2000 IU/mL). We excluded patients with cirrhosis from treatment groups that received 6 weeks of treatment. We assessed the presence or absence of cirrhosis with liver biopsy or with a combination of FibroSURE (LabCorp, Burlington, NC, USA) test plus aspartate aminotransferase to platelet ratio (APRI). Liver biopsy was required in cases of equivocal FibroSURE results. Full eligibility criteria are included in the appendix. We obtained written or oral informed consent from all participants. The study was approved by the institutional review board of the National Institute of Allergy and Infectious Diseases (NIAID) and was done in compliance with the Good Clinical Practice guidelines, the Declaration of Helsinki, and regulatory requirements.
Procedures
Patients were sequentially enrolled into three groups (1:1:1). We contacted patients for screening visits in the order in which patients initially contacted the NIAID study team with interest. We started the study drug in the order in which participants completed eligibility requirements. In the first group, patients were allocated tosofosbuvir and ledipasvir for 12 weeks. In the second group, patients were allocated to sofosbuvir, ledipasvir, and GS-9669 for 6 weeks. In the third group, patients were allocated to sofosbuvir, ledipasvir, and GS-9451 for 6 weeks.
Sofosbuvir (400 mg) and ledipasvir (90 mg) were given as a combination tablet taken once a day. GS-9669 was given as two 250 mg tablets once a day and GS-9451 (80 mg) as one tablet once a day. We enrolled 10 patients per treatment group into a substudy to measure early viral kinetics and pharmacokinetics. Criterion for stopping study drugs was failure to achieve a greater than 2 log10 decline in HCV RNA levels at week 4 after the first dose. Additional criteria were HCV RNA greater than lower limit of quantification after two previous consecutive HCV RNA values less than the lower limit of quantification and greater than a 2 log10 increase in HCV RNA from nadir. The protocol permitted participants who failed treatment the option of treatment with the standard of care, which at the time of the study was pegylated-interferon, ribavirin, and sofosbuvir. This protocol was later amended with an option for retreatment with sofobuvir and ledipasvir for 12 weeks for patients who failed the 6 week treatment regimens. Neither patients nor investigators were masked to treatment allocation.
We measured plasma HCV RNA concentrations using the real-time HCV Assay (Abbott Laboratories, Abbott Park, IL, USA), with a lower limit of quantification of 12 IU/mL and a lower limit of detection of 3 IU/mL. We also measured serum HCV RNA concentrations using the COBAS TaqMan HCV RNA assay, version 2.0 (Roche Diagnostics, Indianapolis, IN, USA), with a lower limit of quantification of 43 IU/mL and a lower limit of detection of 15 IU/mL.
We recorded adverse events and clinical laboratory results throughout the study. Adverse events were graded from one (mild) to four (severe) according to the NIAID Division of AIDS toxicity table (version 1.0). Pill counts were done at several timepoints during treatment.
During the first month of treatment, we measured plasma HCV RNA concentrations at day 0, 1, 3, 5, 7, 10, 14, 21, and 28 in all patients. Data for very early viral kinetics were obtained in a subset of 29 of 60 participants (ten in group one, ten in group two, and nine in group three) by measuring plasma HCV RNA concentrations 0, 1, 2, 4, 8, 12, 24, and 36 h after administration of the first dose of study drugs.
Whole blood was collected using PAXgene Blood DNA tubes (Qiagen, Valencia, CA, USA) and stored at -80°C until DNA extraction was done with use of the PAXgene blood DNA kit (PreAnalytiX, a Qiagen/ Becton Dickinson Company). We established IL28B and IFNL4 genotype for DNA specimens using the 5' nuclease assay with IL28B and IFNL4 allele specific TaqMan probes (ABI TaqMan allelic discrimination kit, Roche Diagnostics, Indianapolis, IN, USA) and the ABI 7500 real-time PCR system (Applied Biosystems, Carlsbad, CA, USA). Genotyping of variants at the rs12979860 (referred to as IL28B genotype) and rs368234815 (IFNL4) loci was done with custom TaqMan assays as previously described.7 We offered an optional liver biopsy sample after treatment completion (within 2 weeks of drug cessation) to all participants who had a pretreatment liver biopsy for staging and who underwent eligibility assessment at the NIH Clinical Center. Histopathological assessments of post-treatment liver biopsies were done by one pathologist (DEK) with liver expertise in a non-blinded fashion at the time of biopsy and was staged according to the Knodell histological activity index.11 We did viral kinetic modelling with a multiscale model12, 13 in all participants who participated in the study (appendix). Deep sequencing of the HCV NS5A and NS5B genes was done by DDL (DDL Diagnostics Laboratory, Rijswijk, Netherlands) in samples collected at baseline and time of virologic failure in patients who relapsed.
Outcomes
The primary endpoint was the proportion of participants with plasma HCV viral load below the lower level of quantification 12 weeks after treatment completion (SVR12). The main safety endpoint was the frequency and severity of adverse events. Secondary endpoints were proportion of participants with unquantifiable HCV viral load at specified timepoints during and after treatment, discontinuations due to adverse events, safety laboratory changes, and the occurence of HCV-resistance mutations. Other uncompleted secondary endpoints are not reported. We also did a post-hoc comparison of viral kinetics between treatment groups. We report outcomes up to 12 weeks, with follow-up of patients up to 48 weeks post-treatment continuing.ll
Statistical analyses
The primary endpoint and the main safety endpoint were based on an intention-to-treat population (all patients who received at least one dose of study drug). We calculated sample size to provide a sufficiently high probability of observing at least one adverse event of probability 10% or more and with prespecified CIs for estimates of efficacy assuming 20 patients in each treatment group. If the true probability of an adverse event due to a regimen was 10% or more, then a sample size of 20 allows an 88% chance of observing at least one such adverse event. With a sample size of 20, if all patients achieved SVR12, then the 95% CI for that estimate is 83-100, and, if 19 patients achieved SVR 12, then the 95% CI for that estimate is 75-100. We compared baseline demographics using the Kruskall-Wallis test for continuous outcomes and χ2 tests for binary outcomes. We compared estimated fall in HCV viral load between groups using a Kruskal-Wallis test and for significant values, multiple comparisons were made between samples using the Conover-Inman14 procedure including correction for multiple tests. Statistical analyses were done with BiAS, Prism 6.0, SAS, STAT-CRUNCH, and S-Plus 8.0.
Role of the funding source
Data collection, review, and analysis were done by NIH investigators. All funders participated in the study design and writing of the report. NIH-affiliated investigators had full access to all data in the study, and AK and the corresponding author had final responsibility for the decision to submit for publication. The Regulatory Compliance and Human Participants Protection Branch of the National Institute of Allergy and Infectious Diseases (NIAID) served as the study sponsor and was involved in the review and approval of the study via the usual peer-review process as well as the study management. The Regulatory Compliance and Human Participants Protection Branch did not have a role in the design of the study, data collection and analysis, interpretation of the data, preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Gilead Sciences provided drug and scientific advice.
Results
Between Jan 11, 2013, and Dec 17, 2013, 73 participants were screened and 60 were enrolled (figure 1). Baseline characteristics were similar between treatment groups (table 1). Participants were mainly black (53, 88%), men (43, 72%), had IL28 non-CC genotype (48, 80%), were infected with HCV genotype 1a (42, 70%), and had high baseline concentrations of plasma HCV RNA (>800 000 IU/mL; 42, 70%). 15 (25%) patients had stage 3 liver disease. Of patients treated with sofosbuvir and ledipasvir, three (15%) had stage 4 disease (table 1), no patients had stage 4 disease in the other groups.
Of the patients allocated to receive sofosbuvir and ledipasvir, all 20 (100%, 95% CI 83-100) had an unquantifiable HCV RNA 12 weeks after completion of treatment�ie, had SVR12. 19 (95%, 75-100) patients allocated to sofosbuvir, ledipasvir, and GS-9669, and 19 (95%, 75-100) patients allocated to sofosbuvir, ledipasvir, and GS-9451, had unquantifiable serum HCV RNA levels 12 weeks after completion of therapy.
One patient (5%) allocated to sofosbuvir, ledipasvir, and GS-9669 for 6 weeks relapsed 2 weeks after completion of treatment. This patient was infected with HCV genotype 1a, had stage 3 liver disease, a baseline HCV viral load of 1 922 287 IU/mL and an IL28B CT genotype. Additionally, 7% of this patient's baseline virus had the M28T NS5A mutant and 13% had the Q30H NS5A mutant. At relapse, a double-mutant M28T and Q30H was detected, which is associated with a more than 1000-fold reduced susceptibility to ledipasvir. Neither sofosbuvir-resistance variant NS5B S282T nor GS-9669-resistance variants were detected in this patient at relapse. The patient took 98% of his drugs, as established by pill counts. One patient treated with sofosbuvir, ledipasvir, and GS-9451 was incarcerated into prison after having SVR after their 4 week follow-visit after completion of therapy. A special session of the NIAID institutional review board deemed that data obtained after incarceration could not be included for publication.
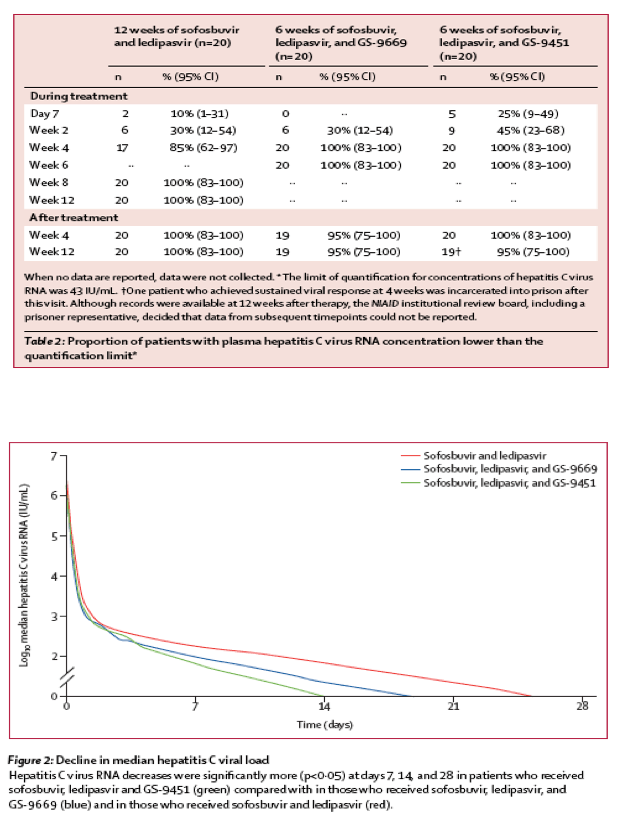
17 (85%) of participants treated with sofosbuvir and ledipasvir had an unquantifiable level (<43 IU/mL of HCV RNA) by week 4 of treatment (table 2). All participants treated with the combination of sofosbuvir, ledipasvir and GS-9669, or that of sofosbuvir, ledipasvir, and GS-9451, had an unquantifiable concentration of HCV RNA by week 4 of treatment (table 2). We noted a rapid, sustained decrease in HCV RNA in patients in all three groups. Viral kinetic modelling shows a three-phase decrease, with a rapid first phase, moderate decay during an intermediate phase, and then a slower third phase (figure 2). This form of decay can be modelled and fitted for every patient (appendix). We assessed whether there was a significantly faster HCV decrease with three drugs compared with two drugs. Fitted HCV RNA concentrations were significantly lower at early timepoints in patients given sofosbuvir, ledipasvir, and GS-9451 than in those given the two other regimens (p<0·05 at days 7, 14, and 21; appendix).
Using our model, we estimated the effect of overall treatment on HCV clearance by taking into account the effectiveness of the regimen in blocking HCV production (δ1 and δ2) and intracellular decay (κ). Median overall treatment effect for all three regimens did not differ significantly (p>0·05; 99·977% for sofusbuvir and ledipasvir, 99·954% for sofosbuvir, ledipasvir, and GS-9669, and 99·984% for sofosbuvir, ledipasvir, and GS-9451; appendix). Nevertheless, more patients in the group given sofosbuvir, ledipasvir, and GS-9451 had a maximum threshold effect of 99·95% or higher in clearance of plasma HCV than did those in the group given the other regimens (p=0·09).
All 60 patients completed treatment. The most common adverse events were diarrhoea, headache, and fatigue (table 3), and most adverse events were mild. Two grade 3 adverse events occurred�pain related to a post-treatment research liver biopsy and an episode of vertigo that caused admission into hospital in a patient who had a history of severe intermittent episodes of vertigo; both patients were allocated to sofosbuvir, ledipasvir, and GS-9451. No grade 4 laboratory abnormalities were reported. 11 grade 3 laboratory abnormalities occurred in nine patients (table 3). Grade 3 hyperglycaemia and hypoglycaemia occurred in one patient who had a history of insulin-dependent diabetes mellitus. Two patients had asymptomatic hypophosphataemia. One patient who had a history of anaemia had transiently decreased haemoglobin concentrations (89 g/L), which improved to baseline concentrations (�100 g/L) before completion of treatment. Three patients had transiently raised serum creatinine concentrations. Two increases occurred after completion of study drugs: one patient with baseline renal insufficiency reported dehydration 8 weeks after treatment (glomerular filtration rate [GFR] 56 mL/min per 1·73 m2 at baseline; 25 mL/min per 1·73 m2 at week 8; 68 mL/min per 1·73 m2 at week 10 without intervention) and another in a patient who initiated 1600 mg per day of ibuprofen for arthritis (GFR 96 mL/min per 1·73 m2 at baseline; 35 mL/min per 1·73 m2 at week 6; 90 mL/min per 1·73 m2 at week 7). A third patient with baseline renal insufficiency had transient worsening of renal function on treatment that resolved without intervention (GFR 66 mL/min per 1·73 m2 at baseline; 35 mL/min per 1·73 m2 at day 7; 59 mL/min per 1·73 m2 at day 9). One patient treated with sofosbuvir and ledipasvir developed an isolated increase in alanine transaminase (peak 94 U/L at week 4) and aspartate aminotransferase (peak 230 U/L at week 5) with normal bilirubin concentrations. Investigations for autoimmune, infectious, and toxin-induced causes had normal findings; however, at week 5, the patient revealed that they had been eating several grapefruits every day for the previous 4 weeks, which has a potential interaction with the atypical antipsychotic lurasidone that the patient was also taking. The patient stopped eating grapefruits and alanine transaminase and aspartate aminotransferase concentrations decreased to pretreatment concentrations by week 6 without interruption of study drug.
Discussion
In our study, patients with chronic HCV genotype 1 infection without cirrhosis were successfully treated with a 6 week course of three oral direct-acting antiviral drugs. The regimens were well tolerated, rapidly suppressed HCV viraemia in patients, and resulted in high rates of SVR12. Treatment for HCV infection is rapidly changing to avoid need for parenteral interferon and oral ribavirin, both of which are associated with many toxic effects, including teratogenicity with ribavirin. Single direct-acting antiviral drugs, including sofosbuvir, were initially used in combination with pegylated-interferon and ribavirin for the treatment of HCV genotype 1 infection, improving efficacy.3, 15, 16 Interferon-free and ribavirin-free regimens have been shown to be efficacious (panel).6, 17, 18, 19, 20 Some interferon-free regimens have been remarkably well tolerated, but the long duration of therapy (12-24 weeks) has raised concerns about adherence and cost.31 Attempts to shorten the duration of therapy to 6 weeks with combination of sofosbuvir, ledipasvir, and ribavirin in a small study resulted in lower SVR rates (68%, 95% CI 47-85) than in patients give 8 weeks (SVR 90-97) or 12 weeks (SVR 92-98) of sofosbuvir and ledipasvir alone.21 Two studies have assessed regimens of 8 weeks' duration.21, 22 In the first study,21 8 weeks of sofosbuvir and ledipasvir was non-inferior to 12 weeks of sofosbuvir and ledipasvir. In the second smaller study,22 8 weeks of ABT-450 and ritonavir, ABT-267, ABT-333, and ribavirin resulted in SVR12 in 86% of patients with HCV genotype 1b infection and in 96% of patients with HCV genotype 1a infection. The investigators did not do a formal comparison of 8 weeks versus 12 weeks with this combination. Although SVR rates were high in both trials, more people relapsed in the groups treated for 8 weeks than in groups treated for 12 weeks, suggesting that efficacy might diminish with these regimens at shorter durations.21, 22 It has not been clear what combinations of antiviral drugs and what duration of therapy are most efficacious, tolerable, and cost effective for patients with a range of host and viral factors.
|
|
| |
| |
|
|
|