| |
Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial
|
| |
| |
Download the PDF
from Jules: these are the phase 2 study results from the LONESTAR study, presented previously at AASLD in the Fall 2013. Since then recently preliminary or top line results were released by Gilead for their Phase 3 studies including larger numbers of patients, over 1,000, across an array of studies, here is link to those results:
GILEAD ANNOUNCES SVR12 RATES FROM THREE PHASE 3 STUDIES EVALUATING A ONCE-DAILY FIXED-DOSE COMBINATION OF SOFOSBUVIR AND LEDIPASVIR FOR GENOTYPE 1 HEPATITIS C PATIENTS - For Immediate Release - (12/18/13)
The Lancet 8 February 2014
Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial
"Patients in the 12-week group of cohort A had a similar response to patients in the 8-week groups, suggesting that 8 weeks of treatment might be sufficient for non-cirrhotic patients who have not previously been treated for HCV......The results in cohort B indicate that 12 weeks of the sofosbuvir plus ledipasvir combination might be an effective treatment for patients who had not achieved sustained virological response with a protease-inhibitor regimen, even in those with compensated cirrhosis......The rate of response in all patients in the study was much the same. In both cohorts, patients with baseline characteristics historically associated with poor response to interferon-based treatment-non-CC IL28b genotype, black race, high baseline viral load-had SVR12 rates similar to patients without those characteristics. Additionally, cohort B included many patients with two other characteristics that have been associated with poor response-over half had compensated cirrhosis and more than two thirds had shown non-response to previous treatment. Rates of SVR12 in these patients were similar to response rates in non-cirrhotic and treatment-naive patients. Overall, only two of the 100 patients treated had virological failure (both relapses), even though nine patients harboured viruses with NS5A baseline variants associated with resistance to treatment. Thus, the presence of these baseline NS5A RAVs (in nine patients) did not preclude the ability of a patient to achieve an SVR12, nor did it accurately predict virological failure."
"In the COSMOS trial,15 in which sofosbuvir was given with the NS3/4A protease inhibitor simeprevir (Janssen Research and Development, Raritan, NJ, USA) with and without ribavirin, sustained virological response rates of 93-96% were reported. In the AI444040 trial,29 investigators treated a broad range of patients-treatment-naive patients with genotypes 1, 2, or 3 infection, as well as patients with genotype-1 infection who had not responded to a previous protease inhibitor regimen-with 12 or 24 weeks of sofosbuvir plus the NS5A inhibitor daclatasvir (Bristol-Myers Squibb, New York, NY, USA). Rates of sustained virological response at 12 weeks after treatment ranged from 86% to 100%.29 The COSMOS trial did not include patients who had not responded to a previous protease inhibitor regimen, whereas the AI444040 trial did not include patients with cirrhosis."
SVR results of a once-daily regimen of simeprevir (SMV, TMC435) plus sofosbuvir (SOF, GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naïve and prior null responder patients: The COSMOS study
http://www.natap.org/2013/AASLD/AASLD_24.htm
EASL/2012: Potent Viral Suppression With the All-Oral Combination of Daclatasvir (NS5A Inhibitor) and GS-7977 (Nucleotide NS5B Inhibitor), +/- Ribavirin, in Treatment-Naive Patients With Chronic HCV GT1, 2, or 3 (100% SVR gt1, 91% gt2) - (04/19/12)
EASL: Sustained Virologic Response With Daclatasvir Plus Sofosbuvir - Ribavirin (RBV) in Chronic HCV Genotype (GT) 1-Infected Patients Who Previously Failed Telaprevir (TVR) or Boceprevir (BOC) - (04/27/13)
Summary
Background
Interferon-based treatment is not suitable for many patients with hepatitis C virus (HCV) infection because of contraindications such as psychiatric illness, and a high burden of adverse events. We assessed the efficacy and safety of an interferon-free regimen-a fixed-dose combination of the nucleotide polymerase inhibitor sofosbuvir (400 mg) and the HCV NS5A inhibitor ledipasvir (90 mg), with and without ribavirin-in patients with genotype-1 hepatitis C infection who were treatment-naive or previously treated with a protease-inhibitor regimen.
Methods
For this open-label study, we enrolled 100 adult patients (>18 years) with HCV infection at a centre in the USA between Nov 2, 2012, and Dec 21, 2012.
In cohort A, we used a computer-generated sequence to randomly assign (1:1:1; stratified by HCV genotype [1a vs 1b])
60 non-cirrhotic, treatment-naive patients to receive -
- sofosbuvir plus ledipasvir for 8 weeks (group 1),
- sofosbuvir plus ledipasvir and ribavirin for 8 weeks (group 2),
- or sofosbuvir plus ledipasvir for 12 weeks (group 3).
In cohort B, we randomly allocated (1:1; stratified by genotype and presence or absence of cirrhosis)
- 40 patients who previously had virological failure after receiving a protease inhibitor regimen to receive
- sofosbuvir plus ledipasvir for 12 weeks (group 4) or
- sofosbuvir plus ledipasvir and ribavirin for 12 weeks (group 5).
22 (55%) of 40 patients in cohort B had compensated cirrhosis.
The primary endpoint was sustained virological response 12 weeks after treatment (SVR12), analysed by intention to treat.
"In cohort A, 53 (88%) of 60 patients had HCV genotype 1a, and 40 (80%) had non-CC IL28B genotypes. A similar proportion of patients in cohort B (34 [85%] of 40 patients) had genotype 1a infection, but a larger proportion (37 patients [93%]) carried non-CC IL28B genotypes, as would be expected given their failure to respond to previous interferon-based treatment combined with a protease inhibitor. More than half of patients (22 patients [55%]) in cohort B had compensated cirrhosis. At baseline, 22 patients with cirrhosis had mean total bilirubin of 0·9 mg/dL, mean albumin of 3·8 g/dL, mean platelet count of 107 x 103, and mean prothrombin time international normalised ratio of 1·27. Most patients in cohort B had shown non-response to previous treatment with protease inhibitor regimens (27 [68%] of 40 patients) whereas about a third (13 patients [33%]) had had virological breakthrough or relapse.
Results
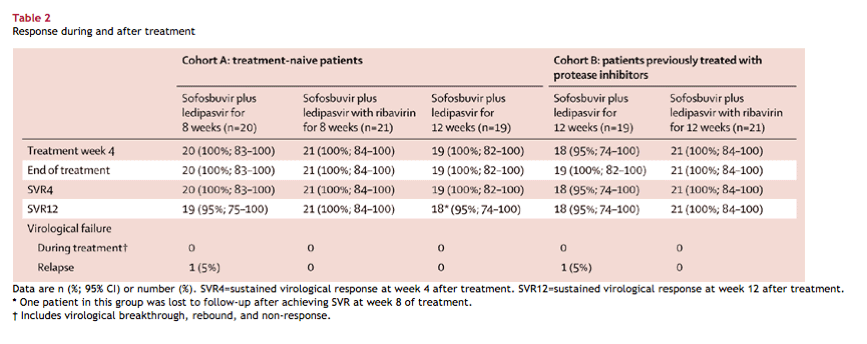
- In cohort A, SVR12 was achieved by 19 (95%) of 20 patients (95% CI 75-100) in group 1,
- by 21 (100%) of 21 patients (84-100) in group 2, and by
- 18 (95%) of 19 patients (74-100) in group 3.
In cohort B, SVR12 was achieved by 18 (95%) of 19 patients (74-100) in group 4
- and by all 21 (100%) of 21 patients (84-100) in group 5.
Two patients had viral relapse; one patient was lost to follow-up after achieving sustained virological response 8 weeks after treatment. The most common adverse events were nausea, anaemia, upper respiratory tract infection, and headache. One patient in group five had a serious adverse event of anaemia, thought to be related to ribavirin treatment.
'No patient in any treatment group had virological breakthrough during study treatment. Of the 100 patients enrolled and treated, two (2%) had virological relapse after receiving a full course of treatment: one non-cirrhotic 60-year-old white man with genotype 1a infection and the CT IL28b genotype in treatment group 1 relapsed between post-treatment weeks 4 and 8, and one cirrhotic 54-year-old white woman with genotype 1a infection and the CT IL28b genotype in treatment group 4 relapsed between post-treatment weeks 2 and 4-this patient was a non-responder to prior treatment with boceprevir-peginterferon-ribavirin triple therapy.
At baseline, NS3 protease inhibitor RAVs were detected in 33 patients: four (7%) of 60 treatment-naive patients and 29 (73) of 40 previously treated patients (table 1). R155K, the most prevalent protease inhibitor RAV, was detected in 19 patients (all of whom were previously treated with protease inhibitors). All 33 patients with baseline NS3 RAVs achieved SVR12.
At baseline, NS5A RAVs were detected in nine patients, seven of whom achieved SVR12. Two patients, one each in groups 1 and 4, had virological relapse as described above. The patient in group 1 had an L31M RAV at baseline; additionally NS5A RAVs (Y93H, Q30L, and L31V) were detected at relapse timepoints. NS5B sequencing also detected the S282T mutation in this patient at relapse. In more than 1900 patients treated to date,20, 21 the S282T mutation, which is associated with reduced susceptibility to sofosbuvir, has been detected once before, in a patient with HCV genotype 2b infection who relapsed after receiving sofosbuvir monotherapy in a phase 2 trial.22 In the second patient who relapsed in this study, the NS5A RAVs Q30H and Y93H were detected at baseline and relapse. The NS5B RAV S282T was not detected in this patient at any post-baseline time point.'
Interpretation
These findings suggest that the fixed-dose combination of sofosbuvir-ledipasvir alone or with ribavirin has the potential to cure most patients with genotype-1 HCV, irrespective of treatment history or the presence of compensated cirrhosis. Further clinical trials are needed to establish the best treatment duration and to further assess the contribution of ribavirin.
Funding
Gilead Sciences.
Introduction
The treatment of patients with chronic hepatitis C virus (HCV) infection has evolved substantially since results from the first clinical trial of recombinant human interferon alfa for this disorder were published in 1986,1 but one element of treatment has remained constant: regular injections of recombinant interferon alfa for durations of 6 months to 1 year. The disadvantages of interferon treatment are well known, from its onerous side-effects (asthenia, myalgia, influenza-like symptoms, cytopenia, and depression) to the inconvenience of weekly injections, and the many potential medical contraindications and other reasons for ineligibility, including the unwillingness of many patients to receive interferon treatment because of side-effects.2, 3 Although protease inhibitor-containing regimens for patients with genotype 1 HCV (both approved in 2011)-24-48 weeks of peginterferon and ribavirin with 12-44 weeks of a protease inhibitor-have achieved high rates of sustained virological response (SVR) in clinical trials, these trials excluded, by definition, the population of patients for whom interferon is not an option because of contraindications or intolerability.4-7 Moreover, HCV protease inhibitors can sometimes cause additional and specific adverse events. In their wider clinical use outside of clinical trials, they have been shown to exacerbate the side-effects normally seen with peginterferon and ribavirin treatment. Serious adverse events associated with these regimens, especially in patients with cirrhosis, can also lead to premature treatment discontinuation.8, 9 Other limitations of protease inhibitors include their low barrier to the development of resistance, high pill burden, complex response-guided regimens, and drug-drug interactions. Overall, less than half of diagnosed patients are eligible to receive protease inhibitor regimens. Those who do not achieve SVR with a protease inhibitor regimen have no treatment options at present.10
Sofosbuvir (formerly known as GS-7977; Gilead Sciences, Foster City, CA, USA) is a nucleotide analogue inhibitor of the HCV NS5B polymerase that has been extensively studied in combination with peginterferon and ribavirin, as well as with other direct-acting antiviral agents in treatment-naive patients with genotype-1 HCV infection.11-15 Ledipasvir (formerly known as GS-5885; Gilead Sciences) is a novel HCV NS5A inhibitor that has shown potent antiviral activity against genotypes 1a and 1b HCV infection,16 and is active against HCV with the S282T mutation, the only variant known to reduce susceptibility to sofosbuvir.17 Findings from a drug interaction study showed no clinically significant pharmacokinetic interactions between sofosbuvir and ledipasvir, with the investigators concluding that the two drugs could be given together without any dose adjustments.18 The combination of sofosbuvir and ledipasvir with ribavirin was first assessed in treatment-naive and prior null responder patients with genotype-1 infection in the ELECTRON trial.19 All 25 treatment-naive patients and all nine prior null responder patients who received 12 weeks of sofosbuvir and ledipasvir plus ribavirin in ELECTRON achieved SVR 12 weeks after discontinuation of treatment (SVR12). However, none of these patients had cirrhosis.
The primary objective of the LONESTAR study was to assess the antiviral efficacy of a fixed-dose single-tablet combination of sofosbuvir and ledipasvir (sofosbuvir plus ledipasvir) with and without ribavirin, for 8 weeks or 12 weeks in treatment-naive patients with genotype-1 HCV, and for 12 weeks in patients with genotype 1 HCV who had not achieved SVR after receiving a protease-inhibitor-containing regimen, half of whom had compensated cirrhosis.
Methods
Study design and participants
For this two-cohort study, we enrolled patients with chronic genotype-1 HCV infection at one clinical site in the USA (Texas Liver Institute, San Antonio, TX). Screening for the trial began on Nov 2, 2012, with the last patient enrolled on Dec 21, 2012. Eligible patients were at least 18 years of age and had chronic genotype-1 HCV infection with serum HCV RNA concentrations of 10 000 IU/mL or greater. Exclusion criteria included hepatic decompensation (as evidenced by the presence of ascites, encephalopathy, or a history of variceal haemorrhage), a body-mass index of 18 kg/m2 or lower, or co-infection with hepatitis B virus or HIV. Patients in cohort A had not received any previous treatment for HCV. Patients in cohort B had had virological failure after treatment with an approved or investigational protease inhibitor regimen (which included peginterferon and ribavirin), but had not discontinued treatment due to an adverse event. About 50% of patients enrolled in cohort B could have compensated cirrhosis. We established the presence of cirrhosis by liver biopsy (eg, a Metavir score of 4 or an Ishak score of ≥5). For all patients, IL28B was determined by PCR amplification and sequencing of the rs12979860 single-nucleotide polymorphism.
Before enrolment and before any study procedures were undertaken, written informed consent was obtained from all patients. The study was done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice.
Randomisation and masking
We used a computer-generated randomisation sequence; allocation to treatment was done sequentially and communicated to the site by email based on the randomisation sequence. XD (Gilead Sciences) generated the randomisation sequence and remained on the study as lead statistician. In cohort A, in which treatment-naive patients with HCV genotype-1 were randomly allocated in a 1:1:1 ratio to treatment groups 1, 2, or 3, randomisation was stratified by HCV genotype (1a vs 1b). In cohort B, in which patients with HCV genotype-1 who had failed a previous protease inhibitor regimen were randomly allocated in a 1:1 ratio to groups 4 or 5, randomisation was stratified by HCV genotype (1a vs 1b) and presence or absence of compensated cirrhosis. This was an open-label study, so patients and investigators were not masked to treatment allocation.
Procedures
Patients in all treatment groups received a fixed-dose combination tablet containing 400 mg of sofosbuvir and 90 mg of ledipasvir (sofosbuvir plus ledipasvir) once daily with or without food. Ribavirin was given orally as a divided, weight-based daily dose (ie, patients weighing <75 kg received 1000 mg and those weighing ≥75 kg received 1200 mg). In cohort A, patients in group 1 received sofosbuvir plus ledipasvir for 8 weeks; those in group 2 received sofosbuvir plus ledipasvir and ribavirin for 8 weeks; patients in group 3 received sofosbuvir plus ledipasvir for 12 weeks. In cohort B, patients in group 4 received sofosbuvir plus ledipasvir without ribavirin for 12 weeks and those in group 5 received sofosbuvir plus ledipasvir with ribavirin for 12 weeks. After completion or early discontinuation of treatment, we followed-up patients for 24 weeks.
We measured plasma HCV RNA concentrations using the COBAS TaqMan HCV Test (version 2.0) for use with the High Pure System (Roche; Indianapolis, IN, USA) with a lower limit of quantification for HCV RNA of less than 25 IU/mL. We defined virological breakthrough as the presence, during treatment, of HCV RNA concentrations greater than 25 IU/mL in serum samples after previous documentation of on-treatment concentrations of HCV RNA concentrations less than 25 IU/mL. We defined relapse as the presence of HCV RNA concentrations greater than 25 IU/mL at any time during the post-treatment follow-up period after documentation of HCV RNA less than 25 IU/mL in a serum sample at the end of treatment.
Deep sequencing of the NS3 and NS5A regions of the HCV RNA was done for all patients at baseline. We established the presence of baseline resistance-associated variants (RAVs) by comparison with wild-type reference sequences (1a-H77 for genotype 1a samples and 1b-Con-1 for genotype 1b samples). NS3 protease RAVs for genotypes 1a and 1b were assessed at aminoacid positions 36, 54, 55, 155, 156, 168, and 170. We also assessed the presence of Q80 polymorphisms. NS5A RAVs was assessed at aminoacids 28, 30, 31, 58, and 93 for genotype 1a, and at positions 31 and 93 for genotype 1b. For all patients who had virological failure, deep sequencing of the NA5A and NS5B regions was done with samples obtained at the time of failure. We compared the resulting sequences with sequences from baseline samples to detect emergent RAVs. We reported RAVs present at greater than 1% of sequence reads.
Patients allocated to groups 1 or 2 who completed 8 weeks of treatment and had post-treatment virological failure at or before the post-treatment week 12 visit were offered a 24-week regimen of sofosbuvir plus ledipasvir with weight-based ribavirin.
We encouraged all patients who achieved SVR 24 weeks after discontinuation of treatment to enrol in the SVR Registry Study (NCT01457755), which is intended to assess durability of SVR for up to 3 years post-treatment. Patients who did not achieve SVR were eligible to enrol in the Sequence Registry Study (NCT01457768), for the purpose of monitoring the persistence of HCV-resistant mutations for up to 3 years post-treatment.
Safety was assessed by review of adverse events and concomitant drugs, blood samples for clinical laboratory testing including haematological assessments, and physical examinations. Patients in groups 2 and 5 (ie, those receiving ribavirin) with decreases in haemoglobin concentrations to lower than 100 g/L during treatment had the option to reduce the daily dose of ribavirin. The use of erythropoiesis-stimulating agents was not permitted.
Statistical analysis
The primary efficacy endpoint of the study was the proportion of patients achieving SVR12, analysed by intention to treat. We did the primary analysis after all patients had been followed-up through 12 weeks post-treatment or after premature discontinuation from the study. The SVR12 rate was calculated for each treatment group along with the 2-sided 95% CI using the exact binomial distribution (the Clopper-Pearson method). The study was exploratory and not powered to allow for comparisons among groups. Therefore, we did no statistical hypothesis testing. With a sample size of 20 patients in each treatment group, a two-sided 95% exact CI will extend at most 46% in length. We used SAS (version 9.2) for all statistical analyses.
This study is registered with ClinicalTrials.gov, number NCT01329978.
Role of the funding source
The study sponsor oversaw trial management, data collection, statistical analyses, and the writing and review of the report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
We screened 116 patients, of whom 100 were deemed eligible and enrolled in the study (figure 1, appendix). Baseline characteristics of patients within each of the two cohorts were similar among groups (table 1). Most patients were non-black. In cohort A, 53 (88%) of 60 patients had HCV genotype 1a, and 40 (80%) had non-CC IL28B genotypes. A similar proportion of patients in cohort B (34 [85%] of 40 patients) had genotype 1a infection, but a larger proportion (37 patients [93%]) carried non-CC IL28B genotypes, as would be expected given their failure to respond to previous interferon-based treatment combined with a protease inhibitor. More than half of patients (22 patients [55%]) in cohort B had compensated cirrhosis. At baseline, 22 patients with cirrhosis had mean total bilirubin of 0·9 mg/dL, mean albumin of 3·8 g/dL, mean platelet count of 107 x 103, and mean prothrombin time international normalised ratio of 1·27. Most patients in cohort B had shown non-response to previous treatment with protease inhibitor regimens (27 [68%] of 40 patients) whereas about a third (13 patients [33%]) had had virological breakthrough or relapse.
All patients had rapid reductions in serum HCV RNA after beginning treatment, with most having serum HCV RNA reductions of about four logs by the end of week 1 of treatment (figure 2). By the end of the second week of dosing, 90 [90%] of 100 patients had HCV RNA below the limit of quantification, and by the end of the fourth week of dosing 99 (99%) of these patients had concentrations below the limit of quantification (appendix).
Most patients in all dose groups achieved SVR12 (table 2), irrespective of previous treatment history (treatment naive vs previously treated), the presence or absence of ribavirin in the regimen, the presence or absence of cirrhosis, or race (black vs non-black). Early viral response during treatment was not predictive of the likelihood of SVR12 achievement. Post-treatment week 24 data are available for all of 97 patients who achieved SVR12. All 97 patients also achieved SVR24.
No patient in any treatment group had virological breakthrough during study treatment. Of the 100 patients enrolled and treated, two (2%) had virological relapse after receiving a full course of treatment: one non-cirrhotic 60-year-old white man with genotype 1a infection and the CT IL28b genotype in treatment group 1 relapsed between post-treatment weeks 4 and 8, and one cirrhotic 54-year-old white woman with genotype 1a infection and the CT IL28b genotype in treatment group 4 relapsed between post-treatment weeks 2 and 4-this patient was a non-responder to prior treatment with boceprevir-peginterferon-ribavirin triple therapy.
At baseline, NS3 protease inhibitor RAVs were detected in 33 patients: four (7%) of 60 treatment-naive patients and 29 (73) of 40 previously treated patients (table 1). R155K, the most prevalent protease inhibitor RAV, was detected in 19 patients (all of whom were previously treated with protease inhibitors). All 33 patients with baseline NS3 RAVs achieved SVR12.
At baseline, NS5A RAVs were detected in nine patients, seven of whom achieved SVR12. Two patients, one each in groups 1 and 4, had virological relapse as described above. The patient in group 1 had an L31M RAV at baseline; additionally NS5A RAVs (Y93H, Q30L, and L31V) were detected at relapse timepoints. NS5B sequencing also detected the S282T mutation in this patient at relapse. In more than 1900 patients treated to date,20, 21 the S282T mutation, which is associated with reduced susceptibility to sofosbuvir, has been detected once before, in a patient with HCV genotype 2b infection who relapsed after receiving sofosbuvir monotherapy in a phase 2 trial.22 In the second patient who relapsed in this study, the NS5A RAVs Q30H and Y93H were detected at baseline and relapse. The NS5B RAV S282T was not detected in this patient at any post-baseline time point.
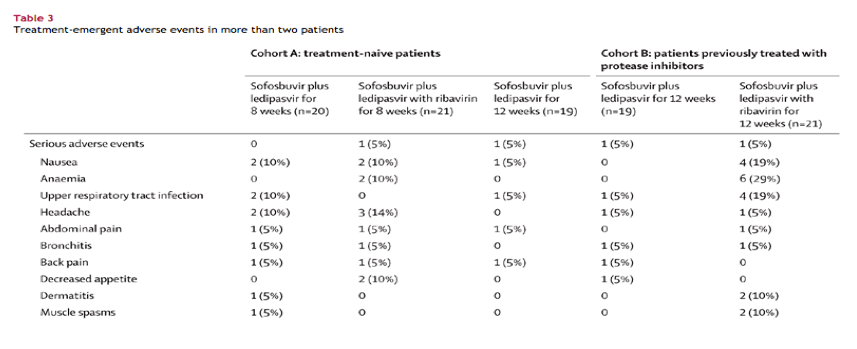
Overall, 48 (48%) of 100 patients had at least one adverse event during the study (table 3). Patients in groups 2 and 5, who were receiving ribavirin with sofosbuvir plus ledipasvir, had the highest rates of adverse events: 12 (57%) of 21 patients in group 2 and 12 (57%) of 21 patients in group 5. Nine (45%) of 20 patients in group 1, eight (42%) of 19 patients in group 3, and seven (37%) of 19 patients in group 4 had an adverse event. The most common adverse events were nausea, anaemia, upper respiratory tract infection, and headache, with most of these events rated by the treating physician as mild in severity. Anaemia was noted only in patients given ribavirin. Eight patients received ribavirin dose reductions to manage anaemia; all eight achieved SVR12. No patient in any group discontinued treatment because of an adverse event. Four patients had serious adverse events: one patient in group 2 had delirium, one patient in group 3 had an exacerbation of peptic ulcer disease, one patient in group 4 was diagnosed with a spinal compression fracture, and one patient in group 5 had anaemia and suicidal ideation; this serious adverse event of anaemia was the only serious adverse event considered related to study treatment. The only grade 3 or 4 haematological abnormality that occurred during treatment was decreased haemoglobin in four patients, all of whom had received ribavirin (two patients in group 2 and one in group 5 had grade 3 reductions in haemoglobin and one patient in group 5 had a grade 4 reduction). In the ribavirin-containing groups, mean change from baseline values in haemoglobin at the end of treatment was -1·8 g/dL in group 2 and -2·0 g/dL in group 5 versus -0·2 g/dL in group 1, 0·0 g/dL in group 3, and -0·2 g/dL in group 4 (figure 3). We recorded no grade 2, 3, or 4 liver chemistry abnormalities in any patient. No patient in any group had abnormal (grade 1-4) increased creatinine concentrations. Patients in the ribavirin-containing groups had mild increases in total bilirubin during treatment (figure 3).
Discussion
In this randomised, open-label study, treatment with the fixed-dose combination of sofosbuvir and ledipasvir with and without ribavirin was well tolerated and resulted in high rates of SVR12 (95-100%) in both treatment-naive and previously treated patients with genotype-1 HCV. Patients in the 12-week group of cohort A had a similar response to patients in the 8-week groups, suggesting that 8 weeks of treatment might be sufficient for non-cirrhotic patients who have not previously been treated for HCV. The results in cohort B indicate that 12 weeks of the sofosbuvir plus ledipasvir combination might be an effective treatment for patients who had not achieved sustained virological response with a protease-inhibitor regimen, even in those with compensated cirrhosis.
The rate of response in all patients in the study was much the same. In both cohorts, patients with baseline characteristics historically associated with poor response to interferon-based treatment-non-CC IL28b genotype, black race, high baseline viral load-had SVR12 rates similar to patients without those characteristics. Additionally, cohort B included many patients with two other characteristics that have been associated with poor response-over half had compensated cirrhosis and more than two thirds had shown non-response to previous treatment. Rates of SVR12 in these patients were similar to response rates in non-cirrhotic and treatment-naive patients. Overall, only two of the 100 patients treated had virological failure (both relapses), even though nine patients harboured viruses with NS5A baseline variants associated with resistance to treatment. Thus, the presence of these baseline NS5A RAVs (in nine patients) did not preclude the ability of a patient to achieve an SVR12, nor did it accurately predict virological failure.
No clinically significant treatment-emergent safety issues were noted in patients receiving the sofosbuvir plus ledipasvir fixed-dose combination. We did not see the haematological abnormalities typically associated with interferon-based treatments, except for the mild anaemia seen in patients receiving ribavirin. The low incidence of adverse events coupled with the brief duration of this regimen contrast favourably with interferon-based treatment, which might mean that this combination treatment could improve treatment adherence and completion compared with interferon-based treatment.
Findings from several phase 2 clinical trials assessing various combinations of direct-acting antiviral agents for 12-24 weeks have established the potential of interferon-free regimens for both treatment-naive and previously treated patients with HCV genotype 1 infection.14,15,23-28 Sofosbuvir has been assessed in combination with other direct-acting antiviral agents. In the COSMOS trial,15 in which sofosbuvir was given with the NS3/4A protease inhibitor simeprevir (Janssen Research and Development, Raritan, NJ, USA) with and without ribavirin, sustained virological response rates of 93-96% were reported. In the AI444040 trial,29 investigators treated a broad range of patients-treatment-naive patients with genotypes 1, 2, or 3 infection, as well as patients with genotype-1 infection who had not responded to a previous protease inhibitor regimen-with 12 or 24 weeks of sofosbuvir plus the NS5A inhibitor daclatasvir (Bristol-Myers Squibb, New York, NY, USA). Rates of sustained virological response at 12 weeks after treatment ranged from 86% to 100%.29 The COSMOS trial did not include patients who had not responded to a previous protease inhibitor regimen, whereas the AI444040 trial did not include patients with cirrhosis. LONESTAR is one of the few early phase trials assessing an interferon-free treatment regimen that includes patients with compensated cirrhosis. In view of the high prevalence of advanced fibrosis in patients with chronic HCV infection and the poor response rates with currently approved therapies, this regimen could fill an important unmet medical need. In terms of safety and efficacy, this regimen compares favourably with other all-oral combinations tested in treatment-naive and previously treated patients with genotype-1 HCV infection and cirrhosis (panel). Other possible advantages of this regimen are its high barrier to viral resistance, short duration, once-daily dosing as a single tablet, absence of food restrictions, few clinically significant drug interactions, and its similar efficacy in genotypes 1a and 1b HCV infections.
-------------------
Panel
research in context
Systematic review
We searched PubMed up to July 22, 2013, using the search term "HCV treatment" for clinical trials for patients with genotype-1 hepatitis C virus (HCV). We also searched the reference list of reviews of the treatment of hepatitis C with direct-acting antiviral agents,30, 31 which systematically surveyed a large body of evidence concerning outcomes of clinical trials.
Interpretation
The standard-of-care regimens for patients with genotype-1 hepatitis C consist of a protease inhibitor-telaprevir or boceprevir-in combination with peginterferon and ribavirin. Among the disadvantages of these regimens are their long duration (24-48 weeks), unfavourable safety and tolerability profiles, poor efficacy in prior null responders with cirrhosis, exclusio n of a large proportion of the patient population because of interferon ineligibility, and complicated regimens with high pill burdens. Many direct-acting agents are in development, and many trials of interferon-free regimens are being done. Data from very few of these trials have been published in peer-reviewed journals to date, but several of these experimental regimens have shown promising efficacy and safety.14,15,23-28 To our knowledge, this trial is the first to report data for cirrhotic genotype-1 patients who had not responded to prior treatment with a protease inhibitor regimen, a population without treatment options at present. Our data lend support to the possibility of effectively treating all patients with genotype-1 HCV with a brief, all-oral, once-daily regimen that has no known safety issues.
--------------------------
The main limitations of this trial are the small size and that it was done at only one centre. This study was not powered to detect small differences in response by characteristics of patients or by treatment groups. Thus, whether 8 weeks of treatment is as efficacious as 12 weeks of treatment or whether ribavirin is necessary in all patients or in only specific populations remain open questions. Three large multicentre phase 3 trials are underway, ION-1 (NCT01701401), ION-2 (NCT01768286), and ION-3 (NCT01851330), and are designed to address the role of treatment duration, the contribution of ribavirin in the regimen, and the effect of compensated cirrhosis in both treatment-naive and previously treated patients. Studies are also planned to assess the sofosbuvir plus ledipasvir fixed-dose combination in patients with more advanced liver cirrhosis (Child-Pugh class B and C), patients with HIV and HCV co-infection, and patients with HCV genotypes 3, 4, 5, and 6.
The results of this trial suggest that the fixed-dose combination of sofosbuvir and ledipasvir could offer a short, all-oral treatment that is effective in both treatment-naive and previously treated patients. Further large scale trials are underway, the findings from which could substantiate those from this trial. Importantly, the sofosbuvir plus ledipasvir combination might be highly effective and safe in patients with multiple negative traditional predictors of response, including cirrhosis, black race, non-CC IL28B variants, and in those who have not responded to standard-of-care protease inhibitor regimens.
|
|
| |
| |
|
|
|