| |
Protein Protects Aging Brain....
new research published....NIH HIV Brain Research Studies
|
| |
| |
Download PDF here
from Jules: Among all too numerous comorbidities that appear to occur more often & earlier among HIV+ individuals is brain, CNS disease. The affects of HIV and a damaged immune system or a dysfunctional immune system appear to be at the vortex of these affects in patients in subclinical & clinical bone, heart, kidney & CNS diseases. Research implicates, and most recently research at CROI 2014 just completed 2 weeks ago in Boston implicates the dysfunctional immune system, and low CD4s and sometimes the presence of HIV viral load, whether it be clearly detectable >50 c/ml, or undetected HIV where it is present but undetectable by standard clinical assay, <50 c/ml. Yes, HIV is present at low levels, below the levels of standard assays, in patients despite having undetectable HIV using standard clinical assays that report HIV <50 c/ml. Often patients have no clinical symptoms of heart disease but may have subclinical heart disease detected by certain tests that measure heart functioning. As well for brain disease, dysfunction may be subclinical. For bone disease one can take a bone dexa test to measure bone loss. But bone disease, low bone mineral density (BMD) is not readily apparent, and can & does simmer below the surface for years, unless identified with a bone dexa. Low BMD is associated with future risk for a bone fracture, and in the general population once a bone fracture occurs in older folks mortality increases. For kidney disease its similar, standard clinical testing has its limits in identifying disease. Clearly monitoring creatinine is a widely used test but it has its limitations. As people age in general kidney disease increases & in HIV+ individuals there appears to be a greater risk for kidney disease. As well brain disease is a concern for HIV+ individuals & all too often it is subclinical, not readily identifiable. CNS or brain disease appears to be more of a "black hole" than these other comorbidities in the sense that there are therapeutic treatments for bone disease, kidney disease, some cancers, & heart disease, although they have limitations. But for brain disease it appears to be less well understood & few if any therapies. Therefore there is a primary need to better research & understand HIV & brain disease.
21st Conference on Retroviruses and Opportunistic Infections
Boston MA
March 3 - 6, 2014
CROI: HCV Coverage at CROI - (03/10/14)
NIH HIV Brain Research Studies:
http://patientinfo.nimh.nih.gov/HIVAIDS_RelatedIssues.aspx#254
http://patientinfo.nimh.nih.gov/HIVAIDS_RelatedIssues.aspx#254
"......in a new study published in Nature, Yankner has found evidence suggesting that the gene that codes for the REST protein switches on again as our brains age [2]. In this biological "second act," REST plays a role in the activation of an anti-stress response that protects aging brain cells from destructive molecules called free radicals and toxic misfolded proteins, such as amyloid, that cause them to degenerate and die."
"There are important implications from this groundbreaking work. First, these lab results suggest that REST may be necessary to keep aging brain cells alive and well. Furthermore, these data suggest that it might be possible to develop ways to restore protective levels of REST, thereby preventing, halting, or maybe even reversing the damage done to neurons in Alzheimer's disease..........
........Yankner's team is now beginning to search for molecules that safely boost REST levels in brain cells. Intriguingly, lithium, which is currently used as a treatment for bipolar illness, raised levels of REST and increased its function in brain cells grown in the laboratory. However, it will take considerably more research to figure out whether this could be a safe and effective treatment for Alzheimer's disease, or whether other drugs that target the REST pathway might be promising.
..........Intriguingly, Yankner's group happened to detect the very highest levels of REST in the brains of people who lived to an extremely old age, such as centenarians. They've also discovered that REST stimulates genes that are known to increase lifespan in animal models. So, as you might imagine, this group of REST researchers won't be resting anytime soon! In addition to all of their work on Alzheimer's disease, they are now keen to explore whether this protein might play an even broader role in healthy aging."
"The researchers are also screening compounds to target REST and other components of its defined chromatin-remodeling complex for drug development purposes........
......."In addition to targeting abnormal proteins in the brain of patients with neurodegenerative diseases, our study suggests that boosting the physiological defense system may also to be important, and that a multipronged therapeutic approach might be beneficial in neurodegenerative disorders, analogous to current cancer therapies," Yanker said."
"When in a person's life are brain cells most vulnerable?" he asked. "The first time is during fetal development, when loss of young neurons would be devastating. The second is during aging, when you're bombarded by oxidative stress and misfolded or aggregated proteins, such as the amyloid beta and tau proteins seen in Alzheimer's disease. It makes sense that a system would come on at those two times to protect neurons, which are largely irreplaceable."
"Having discovered this possible new role for REST, Yankner and team went on to identify the specific genes REST regulates in aging neurons. They found that REST turns off genes that promote brain cell death and contribute to various pathological features of Alzheimer's disease, such as amyloid plaques and neurofibrillary tangles, while it turns on genes that help neurons respond to stress."
"This suggests a person may be able to resist the toxic effects of Alzheimer's pathology if REST levels remain high," said Yankner. "If we could activate this stress-resistance gene network with drugs, it might be possible to intervene in the disease quite early."
------------------------------------
Creative Minds: REST-ling with Alzheimer's Disease
HHS, NIH Blog
http://directorsblog.nih.gov/2014/03/25/creative-minds-rest-ling-with-alzheimers-disease/
Posted on March 25, 2014 by Dr. Francis Collins
Francis S. Collins, M.D., Ph.D., was officially sworn in on Monday, August 17, 2009 as the 16th director of the National Institutes of Health (NIH). Dr. Collins was nominated by President Barack Obama on July 8, and was unanimously confirmed by the U.S. Senate on August 7.
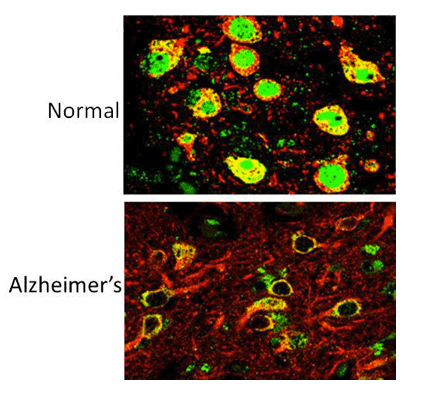
Caption: The REST protein (green) is dormant in young people but switches on in the nucleus of normal aging human neurons (top), apparently providing protection against age-related stresses, including abnormal proteins associated with neurodegenerative diseases. REST is lost in neuron nuclei in critical brain regions in the early stages of Alzheimer's disease (bottom). Neurons are labeled with red.
Credit: Yankner Lab, Harvard Medical School
Why do some people remain mentally sharp over their entire lifetimes, while others develop devastating neurodegenerative diseases that destroy their minds and rob them of their memories? What factors protect the human brain as it ages? And can what we learn about those factors enable us to find ways of helping the millions of people at risk for Alzheimer's disease and other forms of senile dementia?
Those are just a few of the tough questions that Bruce Yankner, a 2010 recipient of the NIH Director's Pioneer Award, has set out to answer by monitoring how gene activity in the brain's prefrontal cortex (PFC) changes as we age. The PFC is the region of the brain involved in decision-making, abstract thinking, working memory, and many other higher cognitive functions; it is also among the regions hardest hit by Alzheimer's disease.
A professor of genetics and neurology at Harvard Medical School in Boston, Yankner's initial work uncovered age-specific changes in activity of certain genes associated with learning and memory [1]. A computational analysis suggested that a master regulator protein, with the awkward name repressor element 1 silencing transcription factor (you can understand why scientists just call this "REST"), may be controlling these genes in older brains. This discovery was surprising, because while REST was known to be active during early fetal development, it was thought that REST was turned off for the rest of a person's life.
Now, in a new study published in Nature, Yankner has found evidence suggesting that the gene that codes for the REST protein switches on again as our brains age [2]. In this biological "second act," REST plays a role in the activation of an anti-stress response that protects aging brain cells from destructive molecules called free radicals and toxic misfolded proteins, such as amyloid, that cause them to degenerate and die.
In their latest work, Yankner and his colleagues compared the levels of REST in a type of brain cell, called a neuron, in the PFCs of young adults (20-35 years) and healthy elderly adults (73-106 years) without Alzheimer's disease. They found that REST was present at low levels in the PFC neurons of young adults, but rose steadily with age. Further examination revealed that REST switches off genes that trigger cell death, along with other genes that have been implicated in Alzheimer's disease.
The researchers went on to look at REST activity in the PFC neurons of people with Alzheimer's disease: some with early and others with late stage disease. It turns out that if a person showed any signs of Alzheimer's disease, REST was almost absent from the control center-the nucleus-of these brain cells. Somehow, REST was evicted from the nucleus and exiled to the cell's "dumpsters," where, along with toxic misfolded proteins, it was destroyed. Similar changes were also seen in the PFC neurons of people suffering from other forms of degenerative brain disease, including conditions such as frontotemporal dementia and dementia with Lewy bodies.
These findings suggest REST may provide a clue to the puzzle of why some older people whose brains contain what's long been considered the hallmark of Alzheimer's disease-amyloid plaques-do not develop the disorder. To investigate this further, Yankner's team tapped into the wealth of clinical information and tissue carefully gathered by the NIH-funded Religious Orders Study, a prospective, long-term human study begun in 1993. In this study, more than 1,000 older members of various Catholic communities have volunteered to undergo annual assessments of their memory and other cognitive skills while they are alive, and, upon their deaths, to donate their brains to research. Yankner and his colleagues focused on the subset of volunteers who had retained strong memory skills until their deaths, despite having brains that showed classic signs of Alzheimer's disease. The researchers' analysis found that these individuals' neurons contained significantly higher levels of REST than did those of their counterparts who had suffered from dementia.
In addition to human studies, Yankner's team explored the function of REST in various model systems, including worms, mice, and brain cells grown in laboratory dishes. Their work showed that when they lowered the activity of the equivalent of the REST gene in the roundworm Caenorhabditis elegans, the microscopic worms were more sensitive to the damaging effects of free radicals and amyloid protein. When the REST gene was added back in, the worms were protected.
Next, the researchers knocked out the REST gene in the brains of mice-a move aimed at mimicking the situation in humans with Alzheimer's disease. When the REST-deficient mice were young, their neurons appeared to be alive and well. However, as these mice grew older, a significant number of neurons died and degeneration set in. In addition, when the investigators removed neurons from the REST-deficient mice and grew them in a lab dish, the brain cells were far more likely to die when exposed to toxic agents, such as free radicals, than those from normal mice. Encouragingly, when researchers inserted the REST gene back into the deficient neurons, they recovered and became more resistant to toxic agents.
There are important implications from this groundbreaking work. First, these lab results suggest that REST may be necessary to keep aging brain cells alive and well. Furthermore, these data suggest that it might be possible to develop ways to restore protective levels of REST, thereby preventing, halting, or maybe even reversing the damage done to neurons in Alzheimer's disease.
Yankner's team is now beginning to search for molecules that safely boost REST levels in brain cells. Intriguingly, lithium, which is currently used as a treatment for bipolar illness, raised levels of REST and increased its function in brain cells grown in the laboratory. However, it will take considerably more research to figure out whether this could be a safe and effective treatment for Alzheimer's disease, or whether other drugs that target the REST pathway might be promising.
Intriguingly, Yankner's group happened to detect the very highest levels of REST in the brains of people who lived to an extremely old age, such as centenarians. They've also discovered that REST stimulates genes that are known to increase lifespan in animal models. So, as you might imagine, this group of REST researchers won't be resting anytime soon! In addition to all of their work on Alzheimer's disease, they are now keen to explore whether this protein might play an even broader role in healthy aging.
-------------------------------------
http://www.the-scientist.com/?articles.view/articleNo/39492/title/Protein-Protects-Aging-Brain/
Protein Protects Aging Brain
Study suggests that REST may be a key regulator of neuronal stress and could play a role in staving off neurodegenerative diseases like Alzheimer's.
By Anna Azvolinsky | March 19, 2014
WIKIMEDIA, NEPHRON
Along with symptoms of cognitive decline, Alzheimer's disease patients often have an accumulation of plaques and tangles of proteins in parts of their brains. But a long-standing question in neurology is why some elderly people develop dementia and others do not. Researchers are also left to wonder why some people have Alzheimer's disease-like brain pathology yet show no cognitive symptoms.
A study published today (March 19) in Nature provides new clues that could help solve both puzzles, showing that a previously unknown stress response kicks in later in life to protect aging neurons. Researchers at Harvard Medical School found that a protein called REST, which is well characterized as a transcription factor that represses neuron-specific genes during embryogenesis, is switched on during middle- and late-adulthood, helping to protect neurons of the hippocampus and cortex from oxidative stress and the aggregated and misfolded proteins characteristic of Alzheimer's and other neurodegenerative diseases.
"This work establishes REST as a regulator protein we have to pay a lot of attention to in the context of neurodegeneration," said Susan Lindquist, a molecular biologist at the MIT Whitehead Institute for Biomedical Research, who was not involved in the study.
Analyzing brain samples from more than 300 individuals who had cognitive function ranging from normal to Alzheimer's disease, the researchers showed that levels of REST were strongly correlated with measures of cognition. Many of the brain samples were from individuals who had died while taking part in a long-term clinical, pathological study and had undergone extensive neuropsychiatric assessments. Tissue from those individuals who retained robust cognitive function at the end of life yet also had Alzheimer's-related plaques had three-fold higher levels of REST in their brains compared to those who had similar pathology but showed signs of dementia.
"This raises the possibility that the structural pathology may not be sufficient to cause Alzheimer's disease," said geneticist Bruce Yankner, who led the work. "A failure of the brain stress response, which REST might mediate, may also be required."
REST first came to the researchers' attention when it emerged among the most active genes in aging human cortices. Using neural cell lines, they next found that REST coordinates the activities of genes that may protect the brain from stress, upregulating the FOXO1 transcription factor-which is known to promote longevity-and binding and repressing cell death-related genes and those that mediate phosphorylation of tau and the generation of amyloid plaques in Alzheimer's disease.
The researchers showed that while primary mouse neurons without REST were highly sensitive to hydrogen peroxide and other stressors, the REST protein could rescue the cell degeneration and cell death phenotype. Mice that lacked REST in their brains showed apoptosis and neurodegeneration in the hippocampus and cortex by their eighth month of life, but not earlier.
The researchers also identified an ortholog of REST in the nematode C. elegans, called SPR-4. SPR-4 mutants showed increased sensitivity to oxidative stress, which could be rescued by expression of either the human REST gene or wild-type SPR-4. When a spr-4 mutant was crossed with the C. elegans model that expresses amyloid-b, the resulting worms exhibited accelerated neurodegeneration. "That REST functions in C. elegans shows this system is likely deeply rooted in evolutionary biology," said Lindquist.
Of course, there are many unanswered questions. Chiefly, whether the REST pathway is a master stress-regulator in neurons-analogous to the heat shock response in other cell types-remains to be seen.
Todd Golde, a neuroscientist and pathologist who studies neurodegenerative diseases at the University of Florida, said the study presents a compelling case that reduction of neuronal REST plays a role in the susceptibility of neurons to stress but cautioned that it doesn't show how exactly the protein is involved in the pathological processes that lead to Alzheimer's disease. "The unanswered question is whether it's a key driver of the degenerative process or just one of many factors that contribute to the downstream degenerative process."
Golde is also somewhat skeptical of the C. elegans and mouse model results. "The experiments in these model organisms show that REST has a role in stress and aging response and can regulate neuronal viability in these systems, but it doesn't really inform on whether REST plays a neuroprotective role in the aging human brain as these are not very good models of human Alzheimer's disease," he said.
Yankner and his colleagues are now working on follow-up experiments to better understand whether REST is indeed a regulator of brain aging. One goal is to isolate the signals that turn REST on in the aging brain, which the researchers suggested could be linked to Wnt signaling. Another is to determine whether REST has a more general stress response function in the context of other neurodegenerative diseases, or in cases of traumatic brain injury or stroke.
The researchers are also screening compounds to target REST and other components of its defined chromatin-remodeling complex for drug development purposes.
"In addition to targeting abnormal proteins in the brain of patients with neurodegenerative diseases, our study suggests that boosting the physiological defense system may also to be important, and that a multipronged therapeutic approach might be beneficial in neurodegenerative disorders, analogous to current cancer therapies," Yanker said.
T. Lu et al. "REST and stress resistance in ageing and Alzheimer's disease," Nature, doi:10.1038/nature13163, 2014.
FULL text, pdf below......
------------------------------------
http://hms.harvard.edu/news/aging-brain-needs-rest-3-19-14
The Aging Brain Needs REST
Research implicates new player in Alzheimer's and other dementias
By STEPHANIE DUTCHEN
March 19, 2014
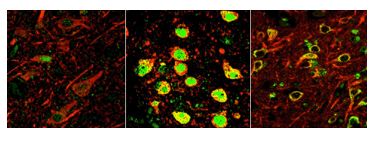
A new study shows that a gene regulator called REST, dormant in the brains of young people (left), switches on in normal aging brains (center) to protect against various stresses, including abnormal proteins associated with neurodegenerative diseases. REST is lost in critical brain regions of people with Alzheimer's (right). Image: Yankner Lab
Why do neurodegenerative diseases such as Alzheimer's affect only the elderly? Why do some people live to be over 100 with intact cognitive function while others develop dementia decades earlier?
More than a century of research into the causes of dementia has focused on the clumps and tangles of abnormal proteins that appear in the brains of people with neurodegenerative diseases. However, scientists know that at least one piece of the puzzle has been missing because some people with these abnormal protein clumps show few or no signs of cognitive decline.
A new study offers an explanation for these longstanding mysteries. Researchers have discovered that a gene regulator active during fetal brain development, called REST, switches back on later in life to protect aging neurons from various stresses, including the toxic effects of abnormal proteins. The researchers also showed that REST is lost in critical brain regions of people with Alzheimer's and mild cognitive impairment.
"Our work raises the possibility that the abnormal protein aggregates associated with Alzheimer's and other neurodegenerative diseases may not be sufficient to cause dementia; you may also need a failure of the brain's stress response system," said Bruce Yankner, Harvard Medical School professor of genetics and leader of the study.
"If true, this opens up a new area in terms of treatment possibilities for the more than 5 million Americans currently living with Alzheimer's disease," said Yankner, who in the 1990s was the first to demonstrate the toxic effects of amyloid beta, the hallmark abnormal protein in Alzheimer's.
The results were published Mar. 19 in Nature.
Protection at the end of life
The CDC lists Alzheimer's disease as the sixth leading cause of death in the United States, and a Mar. 5 paper in Neurology by a group unrelated to Yankner's argued that it should be ranked third. A 2013 study by the RAND Corporation found that with an estimated annual toll of as much as $215 billion, Alzheimer's is America's most expensive disease, costing more than heart disease or cancer.
"Dementia is not an inevitable result of aging," said Yankner, who is also co-director of the Paul F. Glenn Laboratories for Biological Mechanisms of Aging. "We know it's possible for the human brain to work normally for a century or more. So a robust mechanism must have evolved to preserve brain function and keep brain cells alive in long-lived organisms like us. We just haven't learned what that mechanism is."
Yankner believes REST may be a key piece in the solution to that puzzle. REST first came to his attention when team member Tao Lu, HMS instructor in genetics, flagged it as the most strongly activated transcriptional regulator-a switch that turns genes on or off-in the aging human brain. The team confirmed the finding through biochemical and molecular tests and high-resolution imaging.
The finding surprised him at first because until then, REST's only known activity in the brain occurred prenatally, when it keeps key genes turned off until progenitor cells are ready to differentiate into functional, mature neurons. REST was believed to wind down in the brain soon after birth. (It stays active elsewhere in the body and appears to protect against several kinds of cancer and other diseases.) When Yankner thought more about it, however, it began to make sense.
"When in a person's life are brain cells most vulnerable?" he asked. "The first time is during fetal development, when loss of young neurons would be devastating. The second is during aging, when you're bombarded by oxidative stress and misfolded or aggregated proteins, such as the amyloid beta and tau proteins seen in Alzheimer's disease. It makes sense that a system would come on at those two times to protect neurons, which are largely irreplaceable."
Having discovered this possible new role for REST, Yankner and team went on to identify the specific genes REST regulates in aging neurons. They found that REST turns off genes that promote brain cell death and contribute to various pathological features of Alzheimer's disease, such as amyloid plaques and neurofibrillary tangles, while it turns on genes that help neurons respond to stress.
Lab dish experiments revealed that removing REST made neurons more vulnerable to the toxic effects of oxidative stress and amyloid beta. REST appeared to clear away and protect against the free radicals that result from oxidative stress.
To confirm REST's role, the team engineered mice that lacked REST only in their brains and watched what happened as they aged.
"The mice were okay as young adults, but as they got older, neurons in the brain started to die in the same places as in Alzheimer's: the hippocampus and the cortex," said Yankner. "This suggested that REST is essential for neurons to remain alive in the aging brain."
Together with HMS associate professor of genetics Monica Colaiacovo, the team also uncovered a REST equivalent in the tiny worm C. elegans. There, too, the REST equivalent was necessary to protect against free radicals and amyloid toxicity. This suggested the protective function is shared across species.
Diverted from its course
Yankner and colleagues further illuminated the relationship between REST and the aging brain through a combination of lab experiments and studies of brain tissue from elderly people with and without dementia.
The team showed that REST was activated in normal aging brains. The brains of people who developed mild cognitive impairment, by contrast, showed an early decline in REST. The affected brain regions of people with Alzheimer's had hardly any REST left.
"REST loss correlates very closely with memory loss, especially episodic or autobiographical memory, the type that typically declines early in Alzheimer's," said Yankner.
Cell culture experiments suggested REST is activated when stressed neurons send signals to one another, and that once REST is created in a neuron's cytoplasm, it must travel to the nucleus to do its job.
Yankner's group then found that in Alzheimer's, REST gets diverted from its journey to the nucleus, becomes engulfed through a process called autophagy and is eventually destroyed.
The team saw the same striking misplacement of REST when they looked at brain tissue from people with other prevalent neurodegenerative diseases involving dementia, including frontotemporal dementia and dementia with Lewy bodies. In all three dementing illnesses, REST had been swept into the cellular trash bins alongside each disease's abnormal proteins: amyloid beta in Alzheimer's, tau in frontotemporal dementia and alpha-synuclein in Lewy body disease.
"The prevention of REST from getting to the nucleus may be the earliest phase in the loss of REST function. Our laboratory models suggest that this will make neurons much more vulnerable to a variety of stresses and toxic proteins," said Yankner.
Uncovering how REST gets activated and misplaced provides new ideas for how to intercept Alzheimer's. For instance, rather than solely focusing on lowering amyloid beta levels, as clinical trials have done so far without great success, Yankner imagines trying to target REST with drugs such as lithium, which his lab has shown can boost REST function.
REST and dementia-free longevity
Next, Yankner turned to the long-standing puzzle in neurology of how some aging individuals can harbor Alzheimer's disease pathological changes but never become demented.
The team examined brain tissue gathered as part of the Religious Orders Study and the Rush Memory and Aging Project, both funded by the National Institute on Aging. These long-term studies together follow several thousand aging participants and collect donated tissue after death to better understand normal aging, cognitive impairment and neurodegenerative disease.
The team sorted the samples into two groups. One group had Alzheimer's pathology and experienced symptoms of dementia. The second group had the same amount of Alzheimer's pathology but did not become demented. The team found that the group with no dementia had at least three times more REST in the nuclei of their neurons in key brain regions.
"This suggests a person may be able to resist the toxic effects of Alzheimer's pathology if REST levels remain high," said Yankner. "If we could activate this stress-resistance gene network with drugs, it might be possible to intervene in the disease quite early."
"Since Alzheimer's strikes late in life, delaying the onset of disease by just a few years could have a very substantial impact," he added.
In additional studies, the team found that REST strongly correlated with increased longevity. REST levels were highest in the brains of people who lived into their 90s and 100s and remained cognitively intact. Levels stayed high specifically in the brain regions vulnerable to Alzheimer's, suggesting that they might be protected from dementia.
Finally, the team showed that REST increases the expression of several genes known to increase lifespan in model systems of aging.
It remains to be seen how many more pieces will slot in alongside REST in solving the puzzle of aging and dementia. For now, the team's findings offer new ideas for combating a disease that currently has no treatment.
"I'm sure there is something else at play that hasn't been seen or measured yet. REST won't be the end-all. But I think our work will help shift attention to this protective pathway in the aging brain and its role in the prevention of Alzheimer's and other dementing diseases," said Yankner.
"It's a new point of view on the problem."
This study was supported by the National Institutes of Health (Director's Pioneer Award DP1OD006849 and grants P01AG27916, R01AG26651, R01GM072551, P30AG10161, R01AG15819 and R01AG17917) and the Glenn Foundation for Medical Research.
-------------------------
REST and stress resistance in ageing and Alzheimer's disease...... "REST regulates a neuroprotective stress response that may be central to cognitive preservation during ageing."
Nature
(27 March 2014)
"Recent studies suggest that epigenetic regulation of chromatin may modulate the cognitive outcome of a variety of neuropathological states26, 27, 28. It is intriguing that ageing individuals who harbour substantial AD pathology do not appear to progress to dementia when neuronal REST levels are high. This raises the possibility that structural pathology, such as Aß deposition and neurofibrillary tangles, may not be sufficient to cause dementia. Rather, failure of the brain's stress response system may also be necessary, suggesting new possibilities for therapeutic intervention."
Discussion
We have demonstrated a striking induction of the developmental transcriptional repressor REST in specific neuronal populations of the ageing human brain together with epigenetic repression of REST target genes. REST has been investigated extensively as a repressor of neuronal genes during embryonic development, a function that persists in adult non-neuronal cells22. Our findings suggest that REST additionally plays a role as a neuroprotective modulator, in part by repressing genes that promote cell death and the pathology of AD. Moreover, REST increases the expression of FOXO transcription factors that mediate oxidative stress resistance20, as well as the antioxidant enzymes catalase and SOD1, possibly through indirect mechanisms such as repression of micro RNAs. Consistent with these findings, REST confers oxidative stress resistance and protects against toxic insults associated with AD, including Aß oligomers and tau phosphorylation. Furthermore, REST appears to be essential for maintaining neuronal viability in the normal ageing cortex and hippocampus. This protective function is conserved in C.elegans. Previous ChIP-SACO23 and ChIP-chip24 studies also identified REST target genes involved in cell death. In addition, a recent study suggests that REST protects against the toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a model of Parkinson's disease25. Thus, REST may coordinate a stress response that is broadly neuroprotective in the ageing brain.
REST dysfunction may contribute to the pathogenesis of a number of different neurodegenerative disorders. In addition to AD, REST was also significantly depleted in frontotemporal dementia and dementia with Lewy bodies. In each of these disorders, REST was lost from the nucleus and appeared in autophagosomes together with pathological misfolded proteins, including Aß, phosphorylated tau, TDP-43 and α-synuclein. This may represent a common pathogenic mechanism that links altered proteostasis to aberrant gene expression.
Recent studies suggest that epigenetic regulation of chromatin may modulate the cognitive outcome of a variety of neuropathological states26, 27, 28. It is intriguing that ageing individuals who harbour substantial AD pathology do not appear to progress to dementia when neuronal REST levels are high. This raises the possibility that structural pathology, such as Aß deposition and neurofibrillary tangles, may not be sufficient to cause dementia. Rather, failure of the brain's stress response system may also be necessary, suggesting new possibilities for therapeutic intervention.
Abstract
Human neurons are functional over an entire lifetime, yet the mechanisms that preserve function and protect against neurodegeneration during ageing are unknown. Here we show that induction of the repressor element 1-silencing transcription factor (REST; also known as neuron-restrictive silencer factor, NRSF) is a universal feature of normal ageing in human cortical and hippocampal neurons. REST is lost, however, in mild cognitive impairment and Alzheimer's disease. Chromatin immunoprecipitation with deep sequencing and expression analysis show that REST represses genes that promote cell death and Alzheimer's disease pathology, and induces the expression of stress response genes. Moreover, REST potently protects neurons from oxidative stress and amyloid ß-protein toxicity, and conditional deletion of REST in the mouse brain leads to age-related neurodegeneration. A functional orthologue of REST, Caenorhabditis elegans SPR-4, also protects against oxidative stress and amyloid ß-protein toxicity. During normal ageing, REST is induced in part by cell non-autonomous Wnt signalling. However, in Alzheimer's disease, frontotemporal dementia and dementia with Lewy bodies, REST is lost from the nucleus and appears in autophagosomes together with pathological misfolded proteins. Finally, REST levels during ageing are closely correlated with cognitive preservation and longevity. Thus, the activation state of REST may distinguish neuroprotection from neurodegeneration in the ageing brain.
The preservation of cognitive function during ageing has emerged as one of the major medical challenges of the 21st century. A fundamental question is why some individuals age with their cognitive function relatively intact whereas others decline and develop Alzheimer's disease (AD). Early studies suggested that neuronal loss was an integral feature of the ageing brain. With the advent of stereological neuronal quantification, however, it became clear that neuronal cell number is largely preserved in the neocortex and hippocampus of the ageing human brain, declining only in the setting of neurodegenerative disease1, 2, 3. Robust stress-response mechanisms must have evolved, therefore, to preserve neurons and cognitive function across an entire lifespan4, 5.
REST is a repressor of neuronal genes during embryonic development that is downregulated once terminal neuronal differentiation has occurred6, 7, 8. Here we show that REST is induced in the ageing human brain and regulates a network of genes that mediate cell death, stress resistance and AD pathology. This gene network becomes dysregulated at early stages of AD when REST is lost from the nucleus. Conditional REST knockout mice and C. elegans models suggest that REST protects neurons from age-related toxic insults. In ageing humans, elevated REST levels are associated with preservation of cognitive function and increased longevity, even in the presence of AD pathology. Hence, REST regulates a neuroprotective stress response that may be central to cognitive preservation during ageing.
REST is induced in the ageing brain and declines in AD
Transcriptional profiling has demonstrated significant changes in the expression of neuronal genes in the prefrontal cortex of ageing humans9, 10. Analysis of this data set using the Ingenuity Systems IPA platform indicates that the transcription factor most strongly predicted to be activated in the ageing brain is REST (P = 9 x 10-10). Moreover, the 21-base-pair canonical RE1 recognition motif for REST is highly enriched in the age-downregulated gene set (P = 3 x 10-7) (Fig. 1a).
To explore the role of REST in the ageing brain, we measured REST levels in extracts of prefrontal cortex (PFC) from young adult (20-35 years) and aged (73-106 years) individuals without AD. REST expression was significantly increased in the ageing human PFC at both the messenger RNA and protein levels, as determined by quantitative PCR with reverse transcription (qRT-PCR) and western blotting (Fig. 1b, c). Full-length REST was markedly increased; the truncated splice variant REST4 was a minor component, comprising 0.1-0.5% of REST mRNA. Immunofluorescence microscopy using three different antibodies against the amino- or carboxy-terminal domains of REST showed a striking induction of REST in the nucleus of ageing neurons in the PFC and hippocampus (Fig. 1d, e and Extended Data Fig. 1). Much lower levels of REST were detected in microglial cells and astrocytes (data not shown). REST antibody specificity was indicated by ablation of immunoreactivity after antibody preabsorption with a REST blocking peptide, absence of immunoreactivity with matched nonspecific IgG, and loss of immunoreactivity after shRNA-mediated REST knockdown in neural SH-SY5Y cells (Extended Data Fig. 1b, c).
We then investigated whether induction of REST in ageing neurons leads to increased REST-RE1 site binding. To assess REST targeting specifically in neurons, we isolated neuronal nuclei from the PFC by fluorescence-activated cell sorting (FACS) of NeuN-positive nuclei5 (Methods). Chromatin immunoprecipitation (ChIP)-PCR analysis showed a marked induction of REST binding to canonical RE1 motifs in REST target genes in the aged PFC (Fig. 1f). These results indicate that REST expression and function is increased in ageing neurons.
We next examined REST in ageing individuals with mild cognitive impairment (MCI) or AD. REST was almost absent from the nucleus of cortical and hippocampal neurons in AD (Fig. 1d and Extended Data Fig. 1a, d). Punctate cytoplasmic REST immunoreactivity was detected in some AD neurons, but was not detectable in other neurons (Fig. 1d). Loss of nuclear REST was confirmed by quantitative immunocytochemical analysis of 77 cases of sporadic AD compared with 72 age-matched cognitively intact controls, and confirmed by FACS analysis of neuronal nuclei in a subset of cases (Fig. 1e). In 23 out of 24 cases of amnestic MCI, nuclear REST levels were reduced relative to the mean of aged controls (Fig. 1e). A survey of brain regions showed significantly reduced nuclear REST levels in affected neuronal populations in AD, including prefrontal cortical neurons, and neurons of the CA1, CA3 and CA4 subfields of the hippocampus (Extended Data Fig. 1a, d). In contrast, nuclear REST was not reduced in neurons of the dentate gyrus and cerebellum, which are relatively unaffected in AD (Extended Data Fig. 1a, d). Reduced REST levels in AD were confirmed by western blotting of prefrontal cortical grey matter samples, as well as isolated cortical nuclei (Fig. 1c, upper and far right panels, respectively). Furthermore, ChIP-PCR analysis of neuronal nuclei showed significantly reduced or absent REST-RE1 site binding in AD relative to normal ageing controls (Figs 1f and 2a). Thus, REST is induced during normal brain ageing, but is markedly reduced in AD in vulnerable neuronal populations.
REST represses genes involved in cell death and AD
To obtain greater insight into the functional consequences of REST induction in the ageing brain, we explored REST targets by chromatin immunoprecipitation and deep sequencing (ChIP-seq) in the neural cell line SH-SY5Y. REST binding to a subset of the identified target genes was confirmed by ChIP-qPCR (Extended Data Fig. 2). REST ChIP-seq sites were significantly enriched among age-regulated genes in human PFC (P = 1.65 x 10-12), and were predominantly downregulated from the third to the tenth decades of life (Extended Data Fig. 2a). Pathway analysis of the ChIP-seq database showed a highly significant enrichment for genes in cell death pathways and genes that are associated with AD/dementia (Extended Data Fig. 2b, c). Cell death genes include p38 MAP kinase (MAPK11), FAS, FADD, TRADD, BAX, BID, DAXX, PUMA (also known as BBC3), the mitochondrial permeability transition pore ANT1 (also known as SLC25A4) and cytochrome C. REST targets related to the pathology of AD include the genes coding for gamma secretase complex members presenilin 2 and presenilin enhancer 2 implicated in amyloid ß-protein (Aß) generation, and CDK5R1 (also known as p35), CDK5R2 (also known as p39), p38 MAP kinase (also known as MAPK11, 12 and 14) and 14-3-3 zeta (also known as YWHAZ) implicated in tau (also known as MAPT) phosphorylation. REST repressed the expression of these genes in SH-SY5Y cells (Extended Data Fig. 3a, b), and strongly inhibited tau phosphorylation at the AT8 and PHF1 epitopes associated with AD (Extended Data Fig. 3c).
We next examined the regulation of these REST target genes in the normal ageing brain and AD. To distinguish effects at different stages of cognitive decline, we stratified the AD population for least severe (AD1; Mini Mental State Examination (MMSE) score > 18) and most severe (AD2; MMSE score < 8). REST targets related to cell death and AD pathology showed reduced REST binding and elevated mRNA expression at both stages of AD (Fig. 2a, b). REST target genes related to neurotransmission, however, showed elevated mRNA expression in early stage AD1, but reduced expression in later stage AD2 (Fig. 2b). Regression analysis of protein levels showed that nuclear REST is inversely correlated with levels of pro-apoptotic (BAX and DAXX) and AD-related (presenilin 2) proteins in normal ageing and AD neurons (Fig. 2c, d). This was also observed for REST targets associated with neurotransmission (SST and CALB1) during normal ageing and early stage AD1, but not in late stage AD2 (Fig. 2c). The activation-associated histone modification H3K9ac, which is typically reduced by REST, was globally downregulated in normal ageing PFC neurons (Fig. 2e) and was inversely correlated with REST levels (Fig. 2c, d). In contrast, neuronal H3K9ac increased significantly in early AD (Fig. 2e). Thus, loss of nuclear REST in AD is associated with epigenetic derepression of potentially pathogenic genes.
REST is neuroprotective
Because REST represses pro-apoptotic genes, we explored a role in neuroprotection by assessing vulnerability to oxidative stress and oligomeric Aß, stressors associated with brain ageing9 and AD11. Primary cultures of mouse E16 cortical neurons were established from Nestin-Cre:RESTlx/lx (REST cKO) and littermate control embryos. After 1-2 weeks in culture, neuronal differentiation was comparable in REST-deficient and control cultures. Upon treatment with hydrogen peroxide or oligomeric Aß42, REST-deficient neurons showed markedly increased degeneration and cell death relative to controls (Fig. 3a and Extended Data Fig. 4a). REST-deficient neurons were rescued by lentiviral transduction of wild-type REST, leading to expression levels 2-2.5-fold that of wild type (Fig. 3a, Extended Data Fig. 4a). In contrast, a REST mutant that is deficient in nuclear translocation (∼REST)12, 13 was unable to rescue (Fig. 3a, b).
REST-deficient neurons showed increased expression of pro-apoptotic genes in response to oxidative stress and oligomeric Aß (Extended Data Fig. 4b). Thus, endogenous REST is neuroprotective.
REST-deficient mice showed a progressive age-related neurodegenerative phenotype. At 1 month of age, neuronal numbers in the cortex and hippocampus of REST cKO mice were not significantly different from controls. Neuronal degeneration and apoptosis appeared by 8 months of age, as indicated by positive neuronal labelling for cleaved caspase-3 and TdT-mediated dUTP nick end labelling (TUNEL; Fig. 3c, d). This was accompanied by significant neuronal loss in the cerebral cortex and hippocampus, and pronounced gliosis (Fig. 3c, d). These results indicate that REST is essential for maintaining neuronal viability in the ageing brain.
To confirm the findings on REST-deficient neurons, we assessed vulnerability to oxidative stress after REST overexpression or short hairpin RNA-mediated knockdown in neural SH-SY5Y cells. Cell death induced by hydrogen peroxide was significantly reduced when REST was overexpressed by 3- to 20-fold (Extended Data Fig. 4c). Higher levels of REST overexpression increased cell death. REST knockdown by lentiviral shRNA transduction significantly increased H2O2-induced apoptosis in both SH-SY5Y cells and cultured human cortical neurons, which was prevented by shRNA-resistant mouse REST (Extended Data Fig. 4c, d). Furthermore, shRNA-mediated REST knockdown increased levels of reactive oxygen species (ROS), which was prevented by the antioxidant N-acetyl-cysteine (NAC) (Extended Data Fig. 4e, f). Moreover, REST knockdown increased oxidative DNA damage, which was reduced by REST overexpression (Extended Data Fig. 4g). To examine the relationship of REST expression to oxidative DNA damage in the human brain, we double-labelled for REST and the oxidative DNA damage marker 8-oxoguanine (8-oxoG). Linear regression analysis showed a significant inverse relationship between nuclear REST and 8-oxoG levels in normal ageing neurons and in AD (Extended Data Fig. 4h). Thus, REST expression during ageing may reduce oxidative damage.
The nematode Caenarhobditis elegans has served as a model system of ageing and stress resistance4. Two multi-zinc finger transcription factors in C. elegans structurally resemble mammalian REST (spr-3 and spr-4)14, 15, and another gene is closely homologous to CoREST (spr-1)16. To test the role of these genes in oxidative stress resistance, we placed adult worms on nematode growth medium supplemented with paraquat, a superoxide generator. C. elegans lines with mutations in spr-, spr-3 and spr-4 all showed significantly reduced survival relative to wild-type controls (Fig. 4a and Extended Data Fig. 5a). The spr-4(by105) mutant showed the greatest vulnerability. Furthermore, depletion of spr-4 by RNA interference significantly reduced survival in wild-type worms after treatment with paraquat (Extended Data Fig. 5b). SPR-4 was expressed predominantly in neurons (Extended Data Fig. 5c), and was induced by oxidative stress (Extended Data Fig. 5d).
The increased sensitivity of the spr-4 mutant worms to oxidative stress was significantly reduced by transgenic expression of either wild-type SPR-4 or human REST (Fig. 4a, b). To ascertain whether SPR-4 and REST directly modulate ROS levels, worms were labelled with the fluorescent ROS indicator 2',7'-dichlorodihydrofluorescein diacetate (DCFDA). spr-4(by105) mutants treated with paraquat showed significantly elevated ROS levels relative to wild-type, which was reduced by transgenic expression of either wild-type SPR-4 or human REST (Fig. 4c, d). To determine whether REST can substitute for SPR-4 as a transcriptional repressor, we assessed the expression of the presenilin gene hop-1, which is repressed by SPR-4. The expression of hop-1 was elevated in adult spr-4 mutants relative to wild-type worms, and was repressed by both wild-type SPR-4 and human REST (Extended Data Fig. 5e). These results indicate that human REST can functionally substitute for SPR-4 in C. elegans.
SPR-4 also modulated the neurotoxicity of Aß in a C. elegans line that expresses Aß42 in glutamatergic neurons and undergoes age-dependent neuronal loss17. When this Aß-expressing line was crossed with the spr-4(by105) mutant, the resulting line expressed Aß42 in an spr-4 loss-of-function background and showed significantly accelerated neurodegeneration (Fig. 4e). Thus, SPR-4 protects against both oxidative stress and Aß toxicity, consistent with a general role in stress resistance.
Induction of REST by stress and Wnt signalling
o determine whether REST might be induced by oxidative and other forms of stress in the ageing brain, we subjected primary human cortical neuronal cultures to a variety of stressors. Incubation with redox-active iron (Fe+2), hydrogen peroxide, the glutathione synthesis inhibitor buthionine sulphoxide (BSO), or Aß42-induced REST mRNA and protein and increased REST-RE1 site binding (Extended Data Fig. 6a-f). To determine whether secreted factors might contribute to REST induction, cortical neurons were incubated with H2O2 followed by removal of H2O2 and generation of conditioned medium. Addition of stress-related conditioned medium to naive neuronal cultures resulted in a marked induction of REST mRNA expression (Extended Data Fig. 6g). Thus, a variety of stressors induce REST expression, which is mediated at least in part through cell non-autonomous signalling.
To explore the role of cell non-autonomous signalling in the ageing brain, we generated extracts of human PFC from young adult and aged samples and assessed their effects on SH-SY5Y cells. REST was robustly induced by extracts of aged human cortex, but minimally by extracts of young adult cortex (Extended Data Fig. 6h). Furthermore, extracts of AD cortex showed reduced REST-inducing activity relative to age-matched controls (Extended Data Fig. 6h). Because REST has been identified as a target of canonical Wnt-ß-catenin signalling18, we examined the role of this cell non-autonomous signalling pathway. Extracts of aged cortex and conditioned medium from stressed cells treated with H2O2, tunicamycin or wortmannin induced ß-catenin in parallel with REST (Extended Data Figs 6h and 7a, b). Induction of REST and ß-catenin was partially inhibited by the Wnt signalling antagonist Dickkopf (Extended Data Fig. 7a, b). REST was also induced by addition of purified Wnt-3a and Wnt-7a. Moreover, the GSK3-ß inhibitors lithium chloride and Chiron 99021 that activate Wnt signalling induced REST, increased REST-RE1 site binding, and augmented nuclear translocation (Extended Data Fig. 7c-f). To assess Wnt signalling in the human brain, we measured nuclear ß-catenin levels in the PFC of young adult and aged cases. Nuclear ß-catenin was significantly elevated in ageing PFC neurons and colocalized with REST (Extended Data Fig. 7g, h). These results indicate that Wnt-ß-catenin signalling may contribute to the induction of REST in the ageing brain.
Autophagy of REST in neurodegenerative disorders
In AD, loss of REST from the nucleus was often accompanied by localization to punctate cytoplasmic structures that were labelled with the autophagosome markers LC3, ATG7 and ATG12, but not the lysosomal markers LAMP1 and LAMP2 (Extended Data Fig. 8a). To determine if autophagy could alter the nuclear:cytoplasmic distribution of REST, autophagy was activated in SH-SY5Y cells by serum deprivation. This resulted in significantly reduced nuclear REST, which was reversed by treatment with 3-methyladenine or bafilomycin, both inhibitors of autophagy (Extended Data Fig. 8b, c). Because activation of autophagy is characteristic of many age-related neurodegenerative disorders, we asked whether REST is also depleted in other dementing diseases. Examination of cortical sections from patients with frontotemporal dementia (FTD) or dementia with Lewy bodies (DLB) showed marked depletion of nuclear REST (Fig. 5a).
Induction of autophagy in neurodegenerative disorders has been associated with pathogenic misfolded proteins such as Aß (ref. 19). In AD, many cortical and hippocampal neurons showed a notable colocalization of REST with Aß in LC3-positive autophagosomes (Fig. 5b). In contrast, REST did not colocalize with Aß in amyloid plaques. Phosphorylated tau and TDP-43 have been implicated as abnormally folded pathogenic proteins in FTD, whereas α-synuclein has been implicated in DLB. Every case (8) of tau-positive FTD examined showed colocalization of REST with phosphorylated tau ( PHF1 epitope) in LC3-positive autophagosomes (Fig. 5b). Similarly, every case (6) of TDP-43-positive FTD showed colocalization of REST with TDP-43 and LC3 (Fig. 5b). All ten cases of DLB examined showed partial colocalization of REST with α-synuclein in autophagosomes (Fig. 5b). Thus, autophagy of REST, together with pathogenic misfolded proteins, may reduce nuclear translocation in AD and other neurodegenerative disorders.
REST, cognitive preservation and longevity
A central question is whether loss of REST in AD contributes to cognitive decline. We explored this question by using measures of cognitive function derived from neuropsychiatric assessments performed longitudinally in the Religious Orders Study and the Rush Memory and Ageing Project (Methods). Linear regression analysis showed that nuclear REST levels in prefrontal cortical neurons are positively correlated with a measure of global cognition, and separate measures of episodic, semantic and working memory with high levels of statistical significance (Fig. 6a and Extended Data Fig. 9a). Similar results were obtained in pyramidal neurons in the hippocampal CA1 region. For both brain regions, REST levels were most robustly correlated with episodic memory, which typically declines early in the course of AD. Elevated REST levels also predicted reduced AD neuropathology (Fig. 6b and Extended Data Fig. 9b).
We then asked if elevated REST levels might confer resistance to AD pathology. Cases with National Institutes on Aging and Reagan Institute (NIA-Reagan) and Consortium to Establish a Registry for Alzheimer's disease (CERAD) scores indicative of pathological AD, but with mild or no cognitive impairment, were compared to cases with pathological AD together with severe cognitive impairment. Nuclear REST levels were significantly elevated in cognitively preserved cases relative to cognitively impaired cases, despite the fact that both groups met pathological criteria for AD (Fig. 6c). Thus, elevated REST levels are associated with cognitive preservation in individuals who meet pathological criteria for AD.
Because REST is associated with stress resistance and preservation of cognitive function, we explored the relationship to human lifespan. Nuclear REST levels in PFC neurons showed a significant positive correlation with longevity (R = 0.53; P < 10-4) (Fig. 6d). This relationship remained significant after multivariate adjustment for cognitive test scores and indices of neuropathology. Furthermore, the expression of REST target genes showed a highly significant inverse correlation with longevity (Extended Data Fig. 2a). Examination of the neuroanatomical distribution of REST in individuals with extreme longevity showed marked induction in neurons of the PFC and hippocampus, but not in the Purkinje or granule cell neurons of the cerebellum (Extended Data Fig. 10a). Thus, REST expression in specific neuronal populations correlates with longevity.
Discussion
We have demonstrated a striking induction of the developmental transcriptional repressor REST in specific neuronal populations of the ageing human brain together with epigenetic repression of REST target genes. REST has been investigated extensively as a repressor of neuronal genes during embryonic development, a function that persists in adult non-neuronal cells22. Our findings suggest that REST additionally plays a role as a neuroprotective modulator, in part by repressing genes that promote cell death and the pathology of AD. Moreover, REST increases the expression of FOXO transcription factors that mediate oxidative stress resistance20, as well as the antioxidant enzymes catalase and SOD1, possibly through indirect mechanisms such as repression of micro RNAs. Consistent with these findings, REST confers oxidative stress resistance and protects against toxic insults associated with AD, including Aß oligomers and tau phosphorylation. Furthermore, REST appears to be essential for maintaining neuronal viability in the normal ageing cortex and hippocampus. This protective function is conserved in C.elegans. Previous ChIP-SACO23 and ChIP-chip24 studies also identified REST target genes involved in cell death. In addition, a recent study suggests that REST protects against the toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a model of Parkinson's disease25. Thus, REST may coordinate a stress response that is broadly neuroprotective in the ageing brain.
REST dysfunction may contribute to the pathogenesis of a number of different neurodegenerative disorders. In addition to AD, REST was also significantly depleted in frontotemporal dementia and dementia with Lewy bodies. In each of these disorders, REST was lost from the nucleus and appeared in autophagosomes together with pathological misfolded proteins, including Aß, phosphorylated tau, TDP-43 and α-synuclein. This may represent a common pathogenic mechanism that links altered proteostasis to aberrant gene expression.
Recent studies suggest that epigenetic regulation of chromatin may modulate the cognitive outcome of a variety of neuropathological states26, 27, 28. It is intriguing that ageing individuals who harbour substantial AD pathology do not appear to progress to dementia when neuronal REST levels are high. This raises the possibility that structural pathology, such as Aß deposition and neurofibrillary tangles, may not be sufficient to cause dementia. Rather, failure of the brain's stress response system may also be necessary, suggesting new possibilities for therapeutic intervention.
|
|
| |
| |
|
|
|