 |
|
|
| |
Sofosbuvir Plus Velpatasvir (GS-5816) Combination Therapy for Treatment-Experienced Patients With Genotype 1 or 3 Hepatitis C Virus Infection: A Randomized Trial
|
| |
| |
Download the PDF here
Download the PDF here
Ann Intern Med. Published online 10 November 2015
Stephen Pianko, MD, PhD; Steven L. Flamm, MD; Mitchell L. Shiffman, MD; Sonal Kumar, MD; Simone I. Strasser, MD; Gregory J. Dore, MD; John McNally, PhD; Diana M. Brainard, MD; Lingling Han, PhD; Brian Doehle, PhD; Erik Mogalian, PhD; John G. McHutchison, MD; Mordechai Rabinovitz, MD; William J. Towner, MD; Edward J. Gane, MD; Catherine A.M. Stedman, MD; K. Rajender Reddy, MD; and Stuart K. Roberts, MD
"All patients received 12 weeks of treatment that included 400 mg of sofosbuvir once daily.......The SVR12 rates in treatment-experienced patients with genotype 3 HCV infection and cirrhosis who received sofosbuvir plus 100 mg of velpatasvir without and with ribavirin were 88% and 96%......Among patients with genotype 1 HCV infection who had not achieved SVR after previous treatment with a protease inhibitor regimen, SVR12 rates were 100% and 97% in those treated with sofosbuvir plus 25 mg of velpatasvir without and with ribavirin, respectively, and 100% and 96% in those treated with sofosbuvir plus 100 mg of velpatasvir without and with ribavirin. "
"Patients: Treatment-experienced adults with genotype 3 HCV infection without cirrhosis (cohort 1) and with compensated cirrhosis (cohort 2) and patients with genotype 1 HCV infection that was unsuccessfully treated with a protease inhibitor with peginterferon and ribavirin (50% could have compensated cirrhosis) (cohort 3)."
Gilead Submits New Drug Application to U.S. Food and Drug Administration for Fixed-Dose Combination of Sofosbuvir/Velpatasvir for Treatment of All Six Genotypes of Hepatitis C - (11/03/15)
Abstract
Background: Effective treatment options are needed for patients with genotype 1 or 3 hepatitis C virus (HCV) infection in whom previous therapy has failed.
Objective: To assess the efficacy and safety of sofosbuvir plus velpatasvir, with and without ribavirin, in treatment-experienced patients.
Design: Randomized, phase 2, open-label study. (ClinicalTrials.gov: NCT01909804)
Setting: 58 sites in Australia, New Zealand, and the United States.
Patients: Treatment-experienced adults with genotype 3 HCV infection without cirrhosis (cohort 1) and with compensated cirrhosis (cohort 2) and patients with genotype 1 HCV infection that was unsuccessfully treated with a protease inhibitor with peginterferon and ribavirin (50% could have compensated cirrhosis) (cohort 3).
Intervention: All patients received 12 weeks of treatment that included 400 mg of sofosbuvir once daily. Patients in each cohort were randomly assigned to 25 mg of velpatasvir once daily with or without ribavirin or 100 mg of velpatasvir once daily with or without ribavirin.
Measurements: Proportion of patients with sustained virologic response at week 12 after treatment (SVR12).
Results: In cohort 1, SVR12 rates were 85% with 25 mg of velpatasvir, 96% with 25 mg of velpatasvir plus ribavirin, 100% with 100 mg of velpatasvir, and 100% with 100 mg of velpatasvir plus ribavirin. In cohort 2, SVR12 rates were 58% with 25 mg of velpatasvir, 84% with 25 mg of velpatasvir plus ribavirin, 88% with 100 mg of velpatasvir, and 96% with 100 mg of velpatasvir plus ribavirin. In cohort 3, SVR12 rates were 100% with 25 mg of velpatasvir, 97% with 25 mg of velpatasvir plus ribavirin, 100% with 100 mg of velpatasvir, and 96% with 100 mg of velpatasvir plus ribavirin. The most common adverse events were headache, fatigue, and nausea.
Limitation: Treatment assignments were not blinded, and no inferential statistics were planned.
Conclusion: Treatment with 400 mg of sofosbuvir plus 100 mg of velpatasvir for 12 weeks was well-tolerated and highly effective in treatment-experienced patients with genotype 1 or 3 HCV infection.
Primary Funding Source: Gilead Sciences.
---------------------------
Editors' Notes
Context - Options are needed when initial therapy fails for hepatitis C virus (HCV) infection.
Contribution - In a phase 2 trial, treatment-experienced patients with genotype 3 HCV infection who either did not have cirrhosis or had compensated cirrhosis, as well as patients with genotype 1 HCV infection that was unsuccessfully treated with a protease inhibitor plus peginterferon or ribavirin, received sofosbuvir plus 1 of 2 doses of velpatasvir with or without ribavirin. Sustained virologic response rates 12 weeks after treatment were high in all groups that received sofosbuvir plus 100 mg of velpatasvir. Treatment was well-tolerated.
Caution - The number of patients was small.
Implication - Larger trials of sofosbuvir plus velpatasvir are indicated for patients with HCV infection for whom initial therapy fails.
-----------------------------
Of the 6 hepatitis C virus (HCV) genotypes, 1 and 3 are the most common and account for approximately 46% and 22% of all global infections, respectively (1). Chronic infection with genotype 1 HCV is most prevalent in the Americas, Europe, and China, and genotype 3 HCV infection is most prevalent in India, Pakistan, and Southeast Asia (1). With the advent of direct-acting antiviral agents, effective interferon-free combination regimens are now available for most patients chronically infected with genotype 1 or 3 HCV (2, 3). However, some subgroups of patients do not achieve optimal rates of sustained virologic response (SVR) with existing 12-week regimens-in particular, cirrhotic patients with genotype 1 or 3 HCV infection who previously received unsuccessful treatment of HCV infection (4). These patients, who are at increased risk for progression to decompensated cirrhosis, hepatocellular carcinoma, and other liver complications, have a medical need for more effective and well-tolerated treatment (7, 8).
Velpatasvir (Gilead Sciences) is a novel inhibitor of the HCV NS5A protein, which is involved in HCV replication, virion assembly, and modulation of host cellular response. Velpatasvir (formerly GS-5816) has demonstrated potent pangenotypic antiviral activity in vitro (9) and in a 3-day monotherapy study in patients with genotype 1, 2, 3, or 4 HCV infection (10, 11). Pharmacology studies showed no clinically important drug interactions between sofosbuvir and velpatasvir (12). A recent phase 2 study demonstrated the safety and efficacy of 12 weeks of the combination of sofosbuvir and velpatasvir in treatment-naive patients with genotype 1 to 6 HCV infection, with rates of SVR at week 12 after treatment (SVR12) of 93% to 100% (13).
We evaluated the antiviral activity, safety, and tolerability of sofosbuvir administered with 25 or 100 mg of velpatasvir with and without ribavirin for 12 weeks in treatment-experienced patients with genotype 1 or 3 HCV infection. Because previously treated patients have historically had a poorer response than treatment-naive patients, we chose to evaluate sofosbuvir plus velpatasvir with ribavirin, as well as sofosbuvir plus velpatasvir alone.
Methods
Design Overview
This phase 2, multicenter, randomized, open-label study was conducted from June 2013 (when the first patient was enrolled) to August 2014 (when the last patient completed follow-up). The study was originally designed to enroll 2 cohorts of patients with chronic genotype 3 HCV infection who had not achieved SVR after previous therapy with an interferon-based regimen-approximately 100 patients without cirrhosis and 100 patients with compensated cirrhosis. Favorable results in treatment-naive patients with genotype 1 HCV infection in a phase 2 trial of similar design (13) prompted us to amend our protocol to enroll a third cohort of approximately 100 patients with chronic genotype 1 HCV infection who did not achieve SVR after previous therapy with an approved or experimental NS3/4A protease inhibitor in combination with peginterferon and ribavirin. Up to 50% of patients with genotype 1 HCV infection could have compensated cirrhosis.
The protocol was approved by the institutional ethics committees at all sites, and the study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki.
Setting and Participants
The study was conducted at 58 clinical sites: 7 in Australia, 2 in New Zealand, and 49 in the United States. Some patients were recruited partly through a posting of study details on ClinicalTrials.gov and others through referral by their treating physicians.
Adults (aged ≥18 years) with HCV RNA levels greater than 10 000 IU/mL were eligible. The HCV RNA genotype was determined by the central laboratory using the Versant HCV Genotype 2.0 Assay (LiPA) (Siemens). If results were inconclusive, we used the TruGene HCV 5'NC Genotyping Kit (Siemens) with the OpenGene DNA Sequencing System (Siemens). The presence of cirrhosis was established by liver biopsy, a FibroTest score greater than 0.75 and an aspartate aminotransferase-platelet ratio index greater than 2 during screening, or a FibroScan value greater than 12.5 kPa. Exclusion criteria were hepatic decompensation or co-infection with hepatitis B virus or HIV; aminotransferase level more than 10 times the upper limit of normal; direct bilirubin level more than 1.5 times the upper limit of normal; platelet count less than 90 × 109 cells/L; hemoglobin A1c level greater than 8.5%; creatinine clearance rate less than 60 mL/min/1.73 m2, as calculated by the Cockcroft-Gault equation; hemoglobin level less than 11 g/dL for female patients or less than 12 g/dL for male patients; albumin level less than 454.55 μmol/L; and prothrombin time (international normalized ratio) greater than 1.5 times the upper limit of normal. To be eligible, patients must not have achieved SVR after previous treatment of HCV infection and must not have discontinued the previous regimen due to an adverse event. Patients with genotype 3 HCV infection with previous exposure to an approved or experimental HCV-specific direct-acting antiviral agent were excluded. Full eligibility criteria are listed in the study protocol (Supplement). All patients provided written informed consent before screening.
Randomization and Interventions
Three cohorts of eligible patients were enrolled: treatment-experienced patients with genotype 3 HCV infection without cirrhosis, treatment-experienced patients with genotype 3 HCV infection with compensated cirrhosis, and patients with genotype 1 HCV infection whose previous treatment with a protease inhibitor and interferon-based regimen was unsuccessful. Within each cohort, patients were randomly assigned to 4 groups (by a 1:1:1:1 ratio), each of which received 12 weeks of a single oral daily dose of 400 mg of sofosbuvir plus 25 mg of velpatasvir, 25 mg of velpatasvir with ribavirin, 100 mg of velpatasvir, or 100 mg of velpatasvir with ribavirin (Figure). Velpatasvir was orally administered in single daily doses of either 25 or 100 mg. Ribavirin was administered orally in a divided daily dose determined by body weight: 1000 mg daily in patients weighing less than 75 kg and 1200 mg daily in patients weighing 75 kg or greater.
Random assignment of patients was managed with an interactive Web-response system (Bracket). A statistician employed by the sponsor (L.H.) generated the randomization code using SAS, version 9.2 (SAS Institute), which was validated by another statistician employed by the sponsor. The randomization was stratified by cohort. Within cohort 3 (patients with genotype 1 HCV infection), randomization was stratified by genotype 1 subtype (1a or 1b) and cirrhosis status (presence or absence). Investigators (Appendix), patients, and trial personnel were not blinded to treatment assignment.
Outcomes and Follow-up
The primary efficacy outcome measure was SVR12, defined as a serum HCV RNA level below the lower limit of quantification (LLOQ) 12 weeks after completion of treatment. We measured HCV RNA levels with the Cobas TaqMan HCV Quantitative Test, v2.0 (Roche Diagnostics), in combination with the High Pure System Viral Nucleic Acid Kit (Roche Diagnostics), and used an LLOQ of 25 IU/mL.
Secondary efficacy outcome measures included the proportion of patients with virologic failure, which was defined as either on-treatment virologic failure (HCV RNA level at or above the LLOQ after 8 weeks of therapy, confirmed >1 log10 increase in HCV RNA level from nadir, or confirmed HCV RNA level at or above the LLOQ after 2 consecutive HCV RNA levels less than the LLOQ) or relapse (HCV RNA level at or above the LLOQ during the posttreatment period or HCV RNA levels less than the LLOQ at the end of treatment). Other secondary efficacy outcome measures (SVR4, SVR24, and HCV RNA levels less than the LLOQ by study visit and HCV RNA levels and change from baseline in HCV RNA levels through week 12) are not reported here.
The primary safety outcome measure was any adverse event leading to permanent withdrawal of study drugs. Safety assessments during treatment and up to 30 days after treatment included reports of adverse events, standard laboratory testing, 12-lead electrocardiography, assessment of vital signs, and symptom-driven physical examinations. Adverse events were coded using the Medical Dictionary for Regulatory Activities and graded by severity by the investigator according to the Gilead Sciences Grading Scale for Severity of Adverse Events and Laboratory Abnormalities.
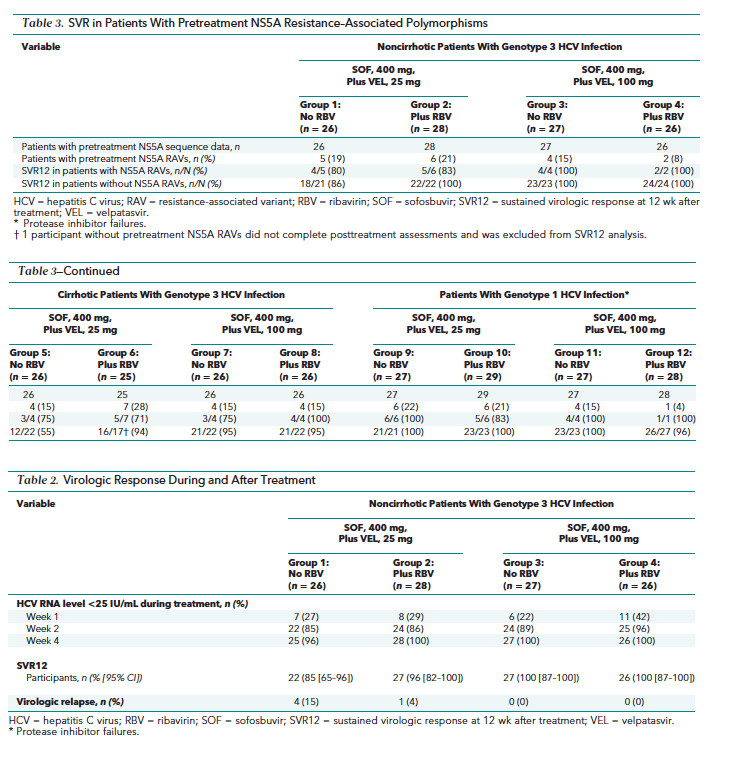
Deep sequencing of the HCV NS5A and NS5B gene was done from pretreatment plasma samples from all enrolled patients and from posttreatment samples from all patients with virologic failure. The HCV NS5A and NS5B coding regions were amplified by DDL Diagnostic Laboratory (Rijswijk, The Netherlands) with standard reverse transcriptase polymerase chain reaction technology, and the reverse transcriptase polymerase chain reaction products were deep sequenced by WuXi AppTec (Shanghai, China). We defined NS5A resistance-associated variants (RAVs) as changes from a genotype-specific reference sequence at amino acid position 24, 28, 30, 31, 32, 38, 58, 92, or 93 (14). We defined NS5B RAVs as any change from the HCV reference sequence at position 96, 142, 159, 282, 289, 320, or 321.
Statistical Analysis
The analysis set for efficacy and safety included randomly assigned patients who received at least 1 dose of a study drug. No inferential statistics or statistical comparison was planned or conducted for efficacy or safety data. A 2-sided 95% CI for the proportion of patients with SVR12 in each treatment group was calculated by the Clopper-Pearson method (15). Patients with missing HCV RNA levels for posttreatment week 12 and after for any reason were counted as having unsuccessful treatment. No formal power or sample size calculations were used to determine group size. With a sample size of 25 patients in each treatment group, a 2-sided exact CI was calculated to extend, at most, 41% in length. We used SAS, version 9.2, for all statistical analyses.
Role of the Funding Source
Gilead Sciences funded this study and was involved in the design, collection, analysis, and interpretation of the data and writing of the report. All coauthors had access to the study data, approved the final report, and assume full responsibility for the veracity of the data and analyses. The lead author had full access to all data and had final responsibility for the decision to submit the manuscript for publication.
Results
Baseline Characteristics
We screened 416 patients, 321 of whom were randomly assigned, received a study drug (Figure and Appendix Table), and were included in the final analysis. Patients were enrolled at 58 sites in the United States, Australia, and New Zealand. The median number of patients enrolled at each site was 5 (range, 1 to 16). Table 1 shows demographic and disease characteristics of the treatment groups. Most patients were men, were white, and had non-CC IL-28B genotype. Approximately one third of patients with genotype 1 HCV infection had cirrhosis. Most patients were classified as having relapse or breakthrough from their previous treatment regimen.
Virologic Response
On-Treatment Response
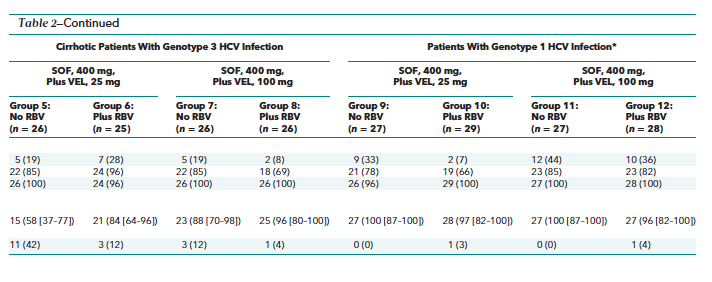
Of the 321 patients who received treatment, 267 (83%) had HCV RNA levels less than the LLOQ by week 2 of treatment; that proportion increased to 99% (318 of 320) by week 4. By the end of treatment, all patients had HCV RNA levels less than the LLOQ, except for 1 patient with genotype 3 HCV infection who received sofosbuvir with 25 mg of velpatasvir and ribavirin (group 6) (Table 2). This patient had HCV RNA levels less than the LLOQ from weeks 2 to 10 but had an HCV RNA level of 43 IU/mL at week 12 and subsequently achieved SVR12.
SVR
Table 2 shows SVR12 rates in all treatment groups. Among the treatment-experienced patients with genotype 3 HCV infection without cirrhosis who received sofosbuvir plus 25 mg of velpatasvir without and with ribavirin, the SVR12 rates were 85% and 96%, respectively. All treatment-experienced patients with genotype 3 HCV infection without cirrhosis who received sofosbuvir plus 100 mg of velpatasvir with or without ribavirin achieved SVR12. All virologic failures in the treatment-experienced patients with genotype 3 HCV infection without cirrhosis were due to posttreatment relapse. One patient discontinued treatment at day 81 due to an adverse event and achieved SVR12.
Among the treatment-experienced patients with genotype 3 HCV infection and cirrhosis who received sofosbuvir plus 25 mg of velpatasvir without and with ribavirin, the SVR12 rates were 58% and 84%, respectively. The SVR12 rates in treatment-experienced patients with genotype 3 HCV infection and cirrhosis who received sofosbuvir plus 100 mg of velpatasvir without and with ribavirin were 88% and 96%, respectively. All virologic failures in the treatment-experienced patients with genotype 3 HCV infection and cirrhosis were due to relapse after treatment. One patient treated with sofosbuvir plus 25 mg of velpatasvir and ribavirin was withdrawn by the investigator for nonadherence to the study protocol and then withdrew consent for subsequent follow-up.
Among patients with genotype 1 HCV infection who had not achieved SVR after previous treatment with a protease inhibitor regimen, SVR12 rates were 100% and 97% in those treated with sofosbuvir plus 25 mg of velpatasvir without and with ribavirin, respectively, and 100% and 96% in those treated with sofosbuvir plus 100 mg of velpatasvir without and with ribavirin. Two patients did not achieve SVR12; both patients, who had genotype 1a HCV infection, had a relapse after the end of treatment. One patient who had a relapse was a white man aged 60 years without cirrhosis who received sofosbuvir plus 25 mg of velpatasvir with ribavirin, and the other was a white woman aged 50 years with cirrhosis who received sofosbuvir plus 100 mg of velpatasvir with ribavirin. No additional relapses were detected in any treatment group after the posttreatment week 12 visit.
Viral Resistance Assessment
Of the 321 patients enrolled, all had pretreatment sequencing data for HCV NS5A and 318 had pretreatment sequencing data for HCV NS5B (Table 3). The prevalence of pretreatment NS5A RAVs detected with 15% cutoff was 17% (53 of 321) overall: 17% (36 of 210) in patients with genotype 3 HCV infection and 15% (17 of 111) in patients with genotype 1 HCV infection. Among patients with genotype 3 HCV infection without cirrhosis, the SVR12 rates were similar in patients with and without NS5A RAVs. Patients with cirrhosis who had genotype 3 HCV infection who were treated with sofosbuvir plus 25 mg of velpatasvir had the lowest overall SVR12 rate at 58% (15 of 26), and only 1 of the 11 patients who had a relapse (9%) (10 of 11 [91%]) had NS5A RAVs (Table 3). Sixteen of 17 (94%) genotype 1-infected patients with pretreatment NS5A RAVs achieved SVR12 (Table 3). The S282T variant was not seen in any patient in this study.
Safety Results
Overall, 263 of 321 patients (82%) had at least 1 adverse event (Table 4). One patient had an adverse event that led to treatment discontinuation. This patient, a white woman aged 58 years without cirrhosis who had genotype 3 HCV infection and received 25 mg of velpatasvir plus ribavirin, had an elevated alanine aminotransferase level (8.75 μkat/L) and γ-glutamyl transferase level (7.55 μkat/L) on treatment day 80. Study treatment was discontinued on day 81. This patient's γ-glutamyl transferase level returned to pretreatment levels by posttreatment day 11 and alanine aminotransferase level normalized by posttreatment day 33. Total bilirubin levels remained normal throughout. The investigator assessed the elevated alanine aminotransferase and γ-glutamyl transferase levels as related to a study drug. This patient achieved SVR12. Across all treatment groups, 8 patients (2%) had serious adverse events that were not considered to be related to a study drug.
Adverse events that occurred in more than 10% of patients across all treatment groups included headache and fatigue (Table 4). More patients in the groups receiving ribavirin had fatigue, nausea, and pruritus; decreased hemoglobin levels; and increased bilirubin levels. One patient had decreased lymphocyte counts and 1 had decreased neutrophil counts that resolved on treatment.
Discussion
The results from this open-label, randomized study suggest that the combination of sofosbuvir and velpatasvir is a safe and effective therapy for treatment-experienced patients with genotype 1 or 3 HCV infection. Although this study was not designed to allow statistical comparisons, especially among subgroups, some preliminary observations may be made about response by velpatasvir dose and the presence or absence of ribavirin.
The SVR12 rates seen in noncirrhotic patients with genotype 3 HCV infection seemed to show a similar pattern of response-those receiving 25 mg of velpatasvir had generally lower rates of SVR12 than those receiving 100 mg. Our data generally suggest that there is no benefit in adding ribavirin to sofosbuvir plus 100 mg of velpatasvir but that ribavirin seems to benefit patients receiving sofosbuvir plus 25 mg of velpatasvir. However, because of the small sample sizes, additional study in larger populations is warranted.
The SVR12 rates seen in treatment-experienced patients with genotype 3 HCV infection receiving 100 mg of velpatasvir compare favorably with those previously reported for other regimens. In the VALENCE study, treatment-experienced patients with genotype 3 without cirrhosis who received 24 weeks of sofosbuvir plus ribavirin had an SVR12 rate of 79%, and those with cirrhosis had an SVR12 rate of 62% (16). In the BOSON study, treatment-experienced patients with genotype 3 HCV infection without cirrhosis who received 12 weeks of sofosbuvir plus peginterferon and ribavirin had an SVR12 rate of 94%, and those with cirrhosis had an SVR12 rate of 86% (17). The ALLY-3 phase 3 study evaluated sofosbuvir plus daclatasvir for 12 weeks in patients with genotype 3 HCV infection. Thirty-two of the 34 (94%) treatment-experienced patients without cirrhosis had SVR12, and 9 of 13 (69%) of those with cirrhosis achieved SVR12 (18).
Patients with genotype 1 HCV infection who had not achieved SVR with previous protease inhibitor regimens had similar rates of SVR12-ranging from 96% to 100%-regardless of velpatasvir dose and presence or absence of ribavirin.
Cirrhosis did not seem to lessen the likelihood of SVR12. Of the 35 treatment-experienced patients with genotype 1 HCV infection and cirrhosis in this cohort, 34 (97%) achieved SVR12. This compares favorably with the results in the ION-2 study among treatment-experienced patients with genotype 1 HCV infection and cirrhosis who had not achieved SVR with a protease inhibitor regimen: SVR12 was achieved by 86% (12 of 14) of those who received 12 weeks of ledipasvir plus sofosbuvir and 85% (11 of 13) of those who received 12 weeks of ledipasvir plus sofosbuvir and ribavirin (4). The populations evaluated in this study and the ION-2 study were similar, although this study had a greater proportion of patients with cirrhosis (31%) and previous unsuccessful protease inhibitor treatment (100%). Approximately 20% of patients in the ION-2 study had cirrhosis, and 61% had a previous unsuccessful protease inhibitor regimen.
The presence of pretreatment NS5A RAVs did not seem to affect SVR12 rates in patients with genotype 1 or 3 HCV infection, except for the patients with genotype 3a HCV infection receiving 25 mg of velpatasvir. The high SVR12 achieved by patients treated with sofosbuvir and 100 mg of velpatasvir with and without ribavirin suggests that this velpatasvir dose effectively suppressed both wild-type virus and virus with RAVs.
Overall, treatment with sofosbuvir and 25 or 100 mg of velpatasvir with or without ribavirin was well-tolerated, with low rates of serious adverse events and only 1 withdrawal due to adverse events. The type and incidence of adverse events or laboratory abnormalities did not suggest toxicity related to velpatasvir dose. Patients who received regimens containing ribavirin had a higher incidence of decreased hemoglobin and elevated bilirubin consistent with ribavirin-induced hemolysis.
This study is limited by its open-label design and because it was not powered for comparisons among treatment groups. In addition, no patients with genotype 2, 4, 5, or 6 HCV infection or decompensated cirrhosis were enrolled in this study. The efficacy of the regimens should not be extrapolated across all HCV genotypes.
In summary, sofosbuvir plus 100 mg of velpatasvir provided high rates of SVR12 in treatment-experienced patients with genotype 1 or 3 HCV infection, including those with compensated cirrhosis, but results will need confirmation in a phase 3 trial. These results support the formulation of a fixed-dose combination tablet of sofosbuvir and 100 mg of velpatasvir for evaluation in phase 3 studies with the goal of developing a simple, all-oral, highly effective, and well-tolerated regimen with broad genotypic coverage.
|
| |
|
|
|
|
|