 |
|
|
| |
HIV Virology and "Cure" Science at CROI 2015
|
| |
| |
by David Margolis MD
UNC Chapel Hill & the Collaboratory of AIDS Researchers for Eradication (CARE)
Forgive me for the use of the word "Cure," now spoken aloud regularly in the context of HIV research and hoped-for future therapy. Here I will try to provide an overview of the presentations that addressed the broad and deepening efforts to develop therapies that might eventually allow a "drug-free remission" of HIV disease. Overall, it seemed to me that the findings presented showed how much the details matter, how complex and difficult the work is, how far we have to go to improve assays, advance current approaches, and develop new ones. But nevertheless, the energy and effort in the field is impressive, and I think bodes well for the future. I have included many hyperlinks in this summary, so that you can link directly to the abstract described, and in many cases to the poster for the presentation, or to other articles of interest. Webcasts of oral presentations can be found here.
The proliferation of cells containing integrated HIV genomes:
A number of presentations highlighted this area of study, recently the subject of several high-profile publications. CD4 T cells that have been infected and contain integrated HIV genomes may undergo cell division, or proliferation. Some of the time, the HIV genomes may be mutated, effectively dead, and functionally irrelevant. Other times the genomes may make virus, but kill the host cell, or the host cell may die as a natural result of terminal proliferation and differentiation. But it may be that some of the time proliferation results in the copying of a latent viral genome, which remains latent in a cell that later returns to the resting state, effectively adding a copy of virus to the latent viral reservoir. How much of the time each of these potential outcomes occurs is the $64,000 question.
Stephen Hughes of the National Cancer Institute (abstr. 21,
webcast) studies recently
published by his group, in parallel with
corroborating studies from a group at U Washington Seattle. The research found that specific integration sites for HIV within the human genome, and that viral integration at these selected locations was linked to clonal expansion and persistence of these cells and the viral genome carried inside them. The work is also summarized in a short Science commentary
Science Commentary] The groups studied the many sites that HIV integrates its DNA into the host genome. Identifying the integration sites in PBMCs and CD4+ T cells from patients identified clonally expanded cells --- cells that encoded exactly the same virus in exactly the same place in more than one cell, a result that can really only be explained by the division of an infected cell, unwittingly copying the HIV genomic integrant as well. Hughes and colleagues identified >2500 integrations, and about 40% of these viral integrations were in clonally expanded cells. In one patient, more than 50% of the infected cells were from a single clone; some of the expanded clones persisted for more than 10 years.
The next surprising and interesting finding was where many of these proviruses were found to land within the genome. Many of the multiple independent integrations were found to be in or near human genes that regulate the growth of cells. This finding suggested that in these cases HIV may have created dysregulation of host cell growth, leading to proliferation of cells that carried copies of the HIV genome.
The potential for virus-induced CD4 T cell proliferation leads to two further questions that are not yet definitively answered. First, could this cell proliferation lead to cancers? While this is possibly so, at least in rare cases, CD4 T cell cancers are not that common in HIV+ patients, and the proliferation of CD4 cells does not explain the increased rates of other kinds of cancers seen in HIV+ patients.
Second, could the proliferation of infected cells expand or preserve the latent reservoir of infection, and drive persistence of HIV infection despite successful ART? While this could be a contributor to viral persistence, as the latent reservoir does not grow over time if there is any proliferation of the persistent pool of virus, it must be matched by the rate at which latently infected cells are spontaneously activated, and release virus. In fact as the latent reservoir slowly (too slowly) is depleted over years of ART, the rate of proliferation must be slightly less than the rate of activation. Further, just before CROI, work published by Nussenzweig at the Rockefeller Institute suggested that the vast majority of these proliferating cells that carry copies of HIV DNA genomes that are defective or dead mutant HIV genomes. This makes a nice story, as one might expect that replication-competent HIV genomes within proliferating cells should make viral particles during proliferation, at least most of the time, and select against the persistence of these viruses. But even a few intact viral genomes that remain in the pool of proliferating cells could contribute to the persistent HIV reservoir.
Finally, the Hughes presentation showed an illustrative case of a single patient in whom the provirus in a single expanded clone was responsible for producing the majority of the virus that was present in the blood of a patient. In this patient, immune surveillance was not sufficient to prevent clonally expanded cells from producing virions. This case illustrated that even defective genomes might produce viral particles that even while defective could still drive immune activation and dysfunction.
Later, Maldarelli, also of the NCI (Abstr. 105,
webcast), presented a detailed, in-depth evaluation of an unusual patient. This unfortunate individual suffered a metastatic squamous cell carcinoma, and was found to have persistent, low-level viremia consisting of a single, clonal viral population. 317 viral sequences were recovered from plasma, PBMC, spleen, lymph nodes, and tumor tissues, which were infiltrated with both CD4+ and CD8+ T cells. This viral clone accumulated specifically in cancer metastases found throughout the patient's body. The proviral clone was recovered and shown not to be the result of the prolonged expression a replication-incompetent mutant virus, but to be able to replicate in PBMCs in culture. These observations suggest that immune stimuli, such as tumor antigens, could contribute to expansion of HIV-infected cells, and perhaps to the activation of the provirus and release of virions into plasma.
Mary Kearney, also of the NCI group, presented detailed studies of clonally expanded viral populations in samples from 4 patients (Abstr. 106,
webcast). Cell-associated HIV RNA (ca-RNA) sequences were extracted from 4-6 separate aliquots of PBMCs using quantitated methods (via internal standards) with high recovery of HIV-1 DNA and RNA. And PCR products were diluted to <1 HIV cDNA molecule/reaction, amplified, and sequenced to obtain ca-RNA Single Genome Sequences (ca-RNA-SGS). An example was shown of patient CAR004, suppressed for 9 years on ART, in whom HIV DNA sequences were diverse, consistent with treatment in the chronic phase of infection following the diversification of the viral pool, but with some isolated clonal expansions of selected ca-RNA-SGS. Expression of these ca-RNA-SGS could be repeatedly found at two times a few weeks apart during ART. Most of these sequences had G>A hypermutants or stop codons, that is they encoded defective RNA messages incapable of producing a virus, but not all were defective and represent some expansion of the reservoir.
Marta Bull presented data with a similar conclusion, performed using different techniques (Abstr. 107,
webcast). 8 patients were studied over 2 years of nevirapine-based ART, based on detection of low-level viremia (LLV; 40-500 copies/mL) after one-year of suppressive (<40 copies/mL) ART. Of note, this is a high threshold of LLV, usually thought of as < 50 c/ml.
Samples were collected every 3 months over the 2-year study. The 8 participants had LLV on 21 of 64 study visits. A median of 21 HIV envelope sequences (range 19-28) were generated from a single LLV, and 90% were identical within a subject. A total of 287 IS sequences were generated from PBMC samples at the LLV visit (median 42 IS/subject, range 27-47).
Proliferation of infected cells, suggested by multiple cells with identical IS, was observed in 6/8 subjects. Two of these six subjects had proliferating clones with env sequences identical to those in LLV.
It will be important to learn how stable the expression of unmutated, potentially replication-competent ca-RNA-SGS is over time, and how many of these cells produce virions. If enough cells with unmutated, potentially replication-competent genomes very often survive the proliferation process and then revert to the latent state, then proliferation could be a significant force that maintains persistent HIV infection.
"Leaky Latency" and measuring HIV RNA, DNA, and viral particles:
A second broad area of work in studies of the mechanisms of persistent HIV infection, efforts to measure the latent reservoir, and to clear is the measurement and characterization of HIV DNA genomes, their HIV RNA products, and their viral offspring.
Ya-Chi Ho (Abstr
392) extended the studies she and colleagues in the SIliciano lab
published in Cell last year. Last year she enumerated that most of the integrated HIV DNA genomes stably within CD4 cells are defective, containing large internal deletions or APOBEC-mediated G-to-A hypermutations. But even if the HIV-1 genome contains lethal mutations, the HIV LTR promoter may remain intact, and cell-associated HIV-1 RNA may still be expressed. This cell-associated RNA (ca-RNA), is the "junk RNA" product of "junk DNA." However, such RNA might still cause problems. It could be translated in to defective HIV proteins, but still cause cellular toxicity or induce an inflammatory immune response. Such defective DNA might be copied if the host cell replicates or proliferates, and thereby foul up efforts to count HIV DNA or its ca-RNA products in attempts to measure the latent HIV reservoir and attempts to deplete it.
So Ho activated resting CD4+ T cells from aviremic ART-treated patients in the lab in the presence of antivirals to prevent new rounds of viral replication in vitro. She asked whether intact or defective HIV-1 can be eliminated by CTLs from those patients. As previously, she found that a significant proportion of the HIV-1 ca-RNA in activated patient CD4+ T cells contains lethal mutations, and that these defective proviruses increased in number over the course of activation, indicating expansion of cells containing defective proviruses upon stimulation. Also, the percentage of defective proviruses increased, implying that at the same time viral cytopathic effect was clearing the reactivated intact proviruses. The amount of HIV-1 proviruses, both intact and defective, decreased after addition of CTLs in some patients, indicating possible elimination by CTLs (http://www.croiconference.org/sites/default/files/posters-2015/392.pdf).
A suggestion that generalized immune stimulation in vivo might turn on viral expression within the latent reservoir was found by Gianella and collegues at UCSD (Abstr. 391). In eleven HIV-infected individuals on suppressive ART (<50 copies/ml) blood samples were obtained at baseline and 1 month after influenza vaccination. Nine of 11 subjects showed an increase in HIV unspliced RNA after influenza vaccination despite undetectable viral loads throughout. Total HIV DNA and 2-LTR circles or HIV RNA did not change, suggesting reactivation of replication-incompetent virus and/or ART-mediated suppression of viral propagation. Potentially, a component of immune stimulation could be considered in the development of eradication strategies.
Another criticism of measurements of HIV ca-RNA has been that some RNAs, most according to the claims of some, could come from transcripts that were initiated upstream or downstream of the viral genome, within a normal host gene, and have simply "read-through" the viral genome. Due to the way a cell reads RNA and turns it in to protein, such accidental, hybrid host-viral RNAs cannot make a normal HIV protein or virus. If this happened most of the time, or even often, measures of ca-RNA (which have become commonly used in cure research) would be poor measures of the state of the latent HIV reservoir. Pasternak from the University of Amsterdam grappled with this problem ( Abstr.
384). He developed a sensitive nested real-time PCR assay that specifically measures host-HIV readthrough transcripts but does not detect genuine HIV-1 unspliced RNA (that could produce a viral particle). He observed only a minor contribution of host-HIV-1 readthrough transcripts to the level of HIV-1 gag RNA. The vast majority of HIV-1 gag RNA transcripts in ART-treated patients represent genuine HIV-1 unspliced RNA. However, it must be remembered, as shown by Ho above, that many of these transcripts may still encode mutations that make them defective.
This possibility was reinforced by the findings of Feiyu Hong from the Mellors laboratory in Pittsburgh (Abstr 379). In a cross-sectional study of 12 viremic patients off ART and of 23 virologically suppressed (<50 cps/ml) patients on ART, PBMCs were tested for total ca-HIV DNA and unspliced HIV-1 RNA using sensitive qPCR. She found a strong, positive correlation between the number of HIV-DNA+ cells and the level of ca-HIV-1 RNA in patients on ART, but no correlation between ca-HIV-1 RNA and the quantity of low-level viremia (measured by single-copy assay). Hong concluded that most of persistent HIV-1 RNA expression in patients on ART does not result in viremia.
In a model system of latency in which cells are infected in the laboratory, DeMaster (Abstr.
390) also found HIV ca-RNA expression in their model of HIV-infected resting CD4+ T cells. Again they found the predominant transcript (usRNA) to be HIV gag. Further they could detect HIV capsid protein in cells from individuals on ART. Although there may be some caveats to this work as some of it was done with cells infected in the laboratory, the findings were consistent with those of others (above). However, as with the other studies shown, the relative contribution to the pool of HIV ca-RNA detected (or even HIV protein in this case) from replication-competent vs. defective proviruses is not known and remains to be carefully documented.
Joseph Casazza (abstr.49webcast) of the NIH Vaccine Research Center presented work using elegant techniques to Identify and characterize single HIV-infected CD4 T cells. He identified broadly-neutralizing antibodies (bNAbs) able to identify individual primary CD4 T cells infected with the laboratory BaL HIV strain by staining for HIV p24 capsid antigen inside cells, in parallel with RNA PCR to detect HIV Gag RNA (full transcripts), and Tat and Rev RNA (short, spliced transcripts). Live cells were then isolated from HIV+ patient's lymph nodes, stained for the exhaustion/activation marker sorted into individual wells, where RNA was extracted, purified, and Gag, and Rev- and Tat-associated RNA copy numbers determined by RT PCR. Measureable Gag-, Tat-, and Rev-associated HIV RNA were assumed to represent active transcription of HIV proviral DNA. Wells were only counted as infected if unspliced Gag was detected, but if so copies of all RNA species were counted. The highest frequency of CD4 T cells transcribing proviral DNA was found in cells that were PD-1+, and either CD4 dim and CD4 negative population. Most of the node CD4 cells counted in this way as expressing HIV RNA were T follicular Helper (Tfh) cells (CD57 and CXCR5 +). In patients not on ART, the highest levels of expression per cell were found in B cell follicle cells. Increased level of Gag p24 antigen detected by the PG121 bnAb was associated with higher levels of Gag RNA, although the association was not tightly linear. In general, the findings provided evidence that the B cell follicle zone of nodes is a highly permissive environment for HIV replication.
Viral rebound off therapy:
Someday, if there has been significant progress towards successful eradication or viral remission therapies, the success of these approaches will face the ultimate test: Analytic Treatment Interruption (ATI). In preparation for that day, some examination of viral rebound off therapy is in order.
Morgane Rolland presented studies (Abstr. 51
, webcast) of the founder populations of HIV viruses enumerated in patients studied during acute HIV infection. In this way, the investigators could tell whether initial HIV infection had been started off by a single virus that crossed the infection threshold and then started to diversify in response to immune pressure, or whether infection was founded by a swarm of different HIV genotypes that had crossed the mucosal threshold and established infection at the same time. In a somewhat chicken-or-egg finding, Rolland discovered that patients who had been infected with multiple founder viruses had significantly higher mean viral loads (0.3 log10 higher), a higher diversity of envelope sequences in the founder population, and in some cases significantly lower CD4 T cell counts over time. It was not clear that the ability to restrict infection and only allow one virus "in the door" led to better outcomes, or if poorer control of infection that allowed multiple viruses in at the same time led to more viral diversity and poorer outcomes.
Jonathan Li (Abstr. 110LB,
webcast) performed a retrospective combined analysis of participants from 5 ACTG studies who were virologically suppressed on ART and received no immunologic intervention prior to undergoing ART was stopped (called analytic treatment interruption, ATI). Li sought markers that could predict time to viral rebound. Rebound was defined as (1) confirmed HIV RNA at or above 200 copies or (2) a single HIV RNA at or above 400 or 1000 copies. They measured ca-HIV RNA, LLV by a single-copy assay, and ca-HIV DNA. Levels of ca-RNA and LLV by 1-copy assay, but not ca-HIV DNA predicted time to viral rebound after ATI. Li stated that "quantification of the active HIV reservoir has the potential to serve as biomarkers of efficacy for therapies that aim to achieve ART-free remission."
Although I appear to be in the minority, I am not excited by this approach to investigating the effect of cure strategies. Certainly, some years from now, cure strategies will have to be tested by an ATI. But the duration of off-ART time in these studies was by and large only a few weeks. I view this as failure, and am not excited about metrics that predict failure. I would not want to test via ATI any cure strategy that did not reduce LLV to stably undetectable. I would like to see a cure strategy reduce ca-HIV RNA. In the future it might be of interest to discover how much a reduction of ca-RNA was correlated with length of aviremic ATI.
John Frater (Abstr. 111LB,
webcast) studied biomarkers for rebound after ATI in SPARTAC study. 18 immunological and virological biomarkers were measured in 154 patients treated in primary HIV-1 infection. 47 participants undertook a TI after 48 weeks of ART. Different than in Li's analysis of ACTG studies, HIV DNA was the only biomarker predictive of during of ATI. There was a trend towards the T cell activation/exhaustion markers PD1, Lag-3 and Tim-3 on CD4 or CD8 cells giving some prediction of time to rebound, but this was not definitive.
Maria Bednar (Abstr. 112LB,
webcast) studied the HIV envelope genes encoded by the first viruses detected in rebound after ATI in ACTG study A5068 in 10 patients. Using single genome amplification (SGA) of the viral env gene, she found low diversity in each subject, consistent with an initially clonal repopulation of the viral population during rebound. No evidence of macrophage-tropic virus was found, as evidenced by the fact that all of the rebound env genes tested encoded proteins that required high levels of CD4 for efficient entry, suggested of so-called T-cell-tropic virus. So in this study, the findings were consistent CD4+ T cells as the source of rebound virus.
Inflammation and HIV infection:
Some have proposed that there is a causal link between mechanisms that drive immune activation in HIV infection, and the persistence of HIV infection on ART. Utay and colleagues at the NIH, Texas, and US Military HIV Research Program found that immune activation persisted even in the face of early initiation of Mega-ART in acute HIV infection (Abstr. 47,
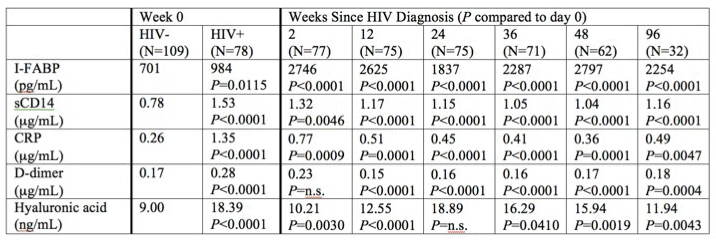
webcast). In the well-known Thai acute HIV cohort (diagnosed with acute HIV infection and initiated ART within 0-5 days per RV254 protocol) 78 volunteers were treated with tenofovir, FTC, efavirenz, and 44 of 78 also received raltegravir, and maraviroc. 20 patients were diagnosed in 4th generation Stage 1 (median 12 days post-acquisition), 15 in Stage 2 (16 days) and 43 in Stage 3 (18 days). HIV RNA levels at the time of treatment were higher in Stage II than earlier. All week 0 biomarker levels were significantly higher in HIV+ than HIV- subjects (see table). Biomarkers of inflammation and immune activation did not decrease until week 12, and some took 24 weeks to decline.
In a parallel analysis of a similar study cohort, Fletcher (abstr. 48, webcast) reported correlation between time to initiate standard ART in acute HIV infection, the detection of HIV RNA in the GALT, and markers of inflammation. Subjects were prospectively enrolled and offered ART during AHI (Fiebig stages I-V) from May 2009 to March 2012 in Bangkok. From 49,458 samples screened for HIV at the Thai Red Cross anonymous clinic, 75 individuals were enrolled during AHI, 41 started ART and 42 consented to optional colon biopsy (so one patient not on ART in this analysis). Sigmoidoscopy was performed to collect colon tissue. Colonic HIV RNA was detectable >50 c/mg in 32 subjects (76%). As compared to subjects without detectable colonic HIV RNA, those with detectable HIV RNA tended to be in a later Fiebig stage (28% Fiebig I in detectable group vs. 80% Fiebig I in undetectable group, p=0.04); had a longer reported duration since HIV exposure; had higher median levels of IP-10, TNF-RII and neopterin; and had higher expression of HLA-DR/CD38 and Ki-67 on CD8 cells in both blood and colon. Total HIV DNA and integrated HIV DNA in the colon correlated with detectable HIV RNA in the colon, as did increases in inflammatory markers. Peripheral CD4 count did not correlate with colonic HIV RNA.
[from Jules: in this interesting study of ART in Acute infection the authors provided this background: the colon is infiltrated during acute HIV infection - may be the site of inoculation, enriched with CD4+ T-cells; suppressive ART started during chronic infection often does not eliminate HIV from the colonic mucosa. Author concluded: detectable HIV RNA is common during AHI & tends to occur during earliest stage of AHI -viral infiltration is broad & rapid. detectable HIV RNA during AHI correlates with: increased HIV burden in the blood, colon & CSF; systemic inflammation; depletion of colonic CD4 cells; CD8+ T-cell activation in the blood & colon. After 24 weeks of ART, many of these differences in viral burden, inflammation & T-cel activation largely disappea
From a therapeutic perspective, Luca Micci (Abstract 168,
webcast) sought evidence that the administration of IL-21, which supports CD4 T cells, might reduce inflammation seen in the SIV infection model. In 16 SIVmac239 chronically-infected rhesus macaques (RMs) 7 months of quadruple therapy given 60 days after infection. Eight RMs received IL-21 for six weeks at the beginning and for 6 weeks at the end of ART, with the other eight serving as ART-treated controls. ART was very effective, with fully suppressed plasma viremia (<60 SIV-RNA copies/ml) in all RMs. Compared to ART-controls, ART+IL-21 RMs showed improved restoration of intestinal Th17 and Th22 CD4+ cells (p<0.01 for both subsets). Further IL-21-treated RMs showed a faster and more pronounced reduction in the levels of activated (HLA-DR+CD38+) and proliferating (Ki-67+) T cells in rectum and blood during ART (p<0.01), and maintained level of T cell activation significantly lower than controls up to eight months following ART-interruption (p<0.01).
Finally, only IL-21-treated animals maintained plasma viral loads significantly lower (from Jules: and reduced immune activation) than those at pre-ART up to eight months post-ART interruption. However, IL-21 is not available for humans now, and could be expected to have some of the effects of IL2. Finally, SIV infection is not always completely predictive of effects seen in HIV. However, IL-21 could provide therapeutic benefits when used as an adjunctive immunomodulatory agent in ART-suppressed HIV-infected individuals.
New therapeutic approaches for persistent HIV infection:
James Whitney (Abstr. 108,
webcast) presented the first in vivo demonstration of a potential new class of latency-reversing agents, in a study of the oral administration of a toll-like receptor 7 (TLR7) agonist in SIV-infected ART-suppressed RMs. The exact mechanism through which TLR agonists are thought to reverse HIV latency is unclear, and direct evidence of the induction of ca-SIV RNA was not presented in this talk. However, TLR7 is present on plasmacytoid dentritic cells and B cells, its signaling increases antigen presentation, can activate CD8 and NK cells to kill target cells, and it can activate CD4 cells. The Gilead TLR7 agonist GS-9620 was developed to augment Hepatitis B virus clearance, via immune action in the liver, and is active against woodchuck and human Hepatitis B in clinical studies. It is extensively metabolized in the gut and the liver, and I understand that systemic exposure is low. This NHP study used an analog of the drug GS-9620. This study was designed to detect latency reversal as measured by the induction of transient plasma viremia or reduction of viral reservoirs. Ten RM were infected with SIVmac251 by rectal challenge. Plasma SIV RNA levels were measured by RT-PCR (limit of detection 50 copies/mL). The RM received ART at ~9 weeks post-infection (PI) and became virologically suppressed by 24 weeks PI; virologic suppression was maintained through week 45. At 45 weeks PI, 4 RM were administered 7 doses of the TLR7 agonist at twice-monthly intervals, while on ART. The first 3 doses were 0.1, 0.2 and 0.3 mg/kg and the last 4 doses remained constant at 0.3 mg/kg. Total viral DNA was quantified in peripheral blood mononuclear cells (PBMC), colon and lymph node biopsies taken pre- and post-completion of TLR7 treatment.
Interestingly, the first 4 doses of TLR7 agonist had no effect on plasma viremia. But doses 4 - 7 led to transient and consistent increases in plasma virus (500 - 1000 SIV RNA copies/mL) in all treated RMs with a return to < 50 copies/mL 4-7 days later. Although the data is sparse, there appeared to be a ceiling effect, in that no viral blips were >1000 c/ml. Surprisingly, after completion of the TLR7 regimen, SIV DNA levels were reduced by 56-75% in PBMC, colon and lymphoid tissues. Viral DNA levels remained unchanged in the placebo control RM. ART was then discontinued, and the TLR7 treated animals showed a ~0.5 log10 reduction in plasma virus setpoint as compared to the placebo group, although the time-to-rebound appeared unchanged.
The agonist had immune effects as well. CD8, CD56 (NK), and to some extent CD4 cells displayed increased levels of the activation marker CD69 for 3 days after each dose. Virologically, the plasma virus isolated during blips had identical sequences to that amplified from PBMC, lymph nodes, and colon. Hypermutant proviral sequences were not found in blips.
Overall the presentation was exciting and interesting, but raised a host of questions. The monkey were very well-suppressed, but it takes a long time to fully dampen smoldering SIV or HIV infection, and so it will be important to know if viremia was induced by virus leaving latency in resting CD4 cells, or by the induction of viral expression in other cell populations. It will also be important to know if this effect was induced directly on latent resting CD4 reservoirs, or by some other effect of immune activation more broadly. It will be important to understand if this was a local effect in the gut and liver, or if reservoirs throughout the animal were effected. The reduction seen in SIV DNA was surprising, and has not thus far been seen with other latency-reversing strategies. It has been postulated that other such strategies (eg HDAC inhibitors) reverse latency to do not kill or clear latently infected cells. If infection is being cleared by TLR7 agonists, it will be important to understand how clearance is being effected.
Sarah Palmer then presented an in-depth viral analysis of samples from the panobinostat HDAC inhibitor study published by Rasmussen (
Abstr. 109,
webcast). The Palmer laboratory sought to determine if the increase in unspliced cell-associated RNA (CA RNA) were due to transcription from a subset or a broad range of latent proviruses. Single-genome sequencing of the env region was used to characterize the virus from the ca-HIV DNA and ca-HIV RNA and plasma HIV RNA. The sequences obtained from the preART plasma reflected the infection status of the patient: limited diversity in acutely-treated patients, more diversity in chronically treated ones. Phylogenetic analysis revealed that the panobinostat-induced plasma viral RNA intermingled extensively with the ca-DNA sequences from the equivalent time points, as would be seen if panobinostat activated transcription from a broad range of proviruses. The rebound virus from the ATI plasma was composed of expansions of homogenous sequences, similar to the panobinostat-induced ca-RNA sequences and ca-DNA sequences. So overall, the data was consistent with panobinostat induction of expression and viral production in a broad range of integrated viruses.
But this data could not rule out the possibility that some viruses were resistant to the effect of panobinostat. Unfortunately, this could only be proven by another inducer that revealed such viruses. Also, 38% of ca-RNA viral sequences were not replication competent, showing that panobinostat may induce both competent and mutated HIV genomes. Overall, the data was encouraging for the efficacy of panobinostat induction, but also illustrated the challenges inherent in attempting to fully clear the latent reservoir.
Several other types of novel therapeutic approaches were presented. The NIH Vaccine Research Center group presented findings illustrating the potent antiviral potential of bNAbs in SHIV-infected macaques (Abstr. 50,
webcast). Rhesus macaques were infected with a low-dose challenge of SHIV-SF162P3 (20 TCID 50) followed by administration 10 days later of either 1) daily triple drug ART; 2) a single dose of Env CD4-binding site specific bNAb, VRC01; 3) a single dose of a combination of a more potent clonal relative of VRC01 (VRC07-523) and a V1/V2 glycan-dependent bNAb (PGT121); or 4) no treatment. Daily ART with FTC/PMPA/RAL was initiated 11 days after bNAb (total bNAb mix dose 20 mg/kg). A single infusion of VRC01 on day 10 reduced viremia by ~ 1 log10 over the next 10 days. The combination of VRC07 and PGT121 had a greater effect of between 2 and 3 log10 that was similar to treatment with ART. Following peak viremia, control was better sustained by the dual bNAb and ART regimens than the VRC01 regimen. Greater efficacy of VRC07-523 / PGT121 relative to VRC01 is consistent with more potent neutralization activity of these bNAbs against the SHIV-SF162P3 challenge virus. However, overall HIV DNA in LN cells, total, naïve, CM, and Tfh cells were decreased by regimens, but in the end there was no difference between treatments. These data illustrate the antiviral potency of bNAbs, and support future therapeutic clinical trials.
Michael Farzan (abstract 163,
webcast) presented his study recently published in Nature (pdf attached, see at top for download)
illustrating the ability of a synthetic antibody-like molecule produced in muscle cells infected with a gene therapy viral vector called AAV could protect RMs from SHIV-AD8 challenges. The genetic delivery of vectors in to cells to chronically produce HIV-preotective antibodies has been proposed and modeled in the past, but this is an impressive new use of AAV-driven gene delivery technology. Farzan showed that the syntheitic antibody-like molecule called eCD4-Ig bound strongly to the HIV-1 envelope and was highly potent. Because eCD4-Ig only binds conserved regions of the Env, it may be more effective than natural broadly neutralizing Abs. In monkeys, the gene therapy construct expressed synthetic antibody for 3-5 years, reaching titers of 15-75 μg/ml in primates. The long term risk of the delivery and production of a foreign antibody needs to be assessed. Farzan suggested that a genetic "off" switch to stop Ab production when it was not needed, might be required to allow this approach to be developed clinically. Obviously, this would be a powerful tool for prevention in an at-risk population, if it could be delivered affordably, and might have roles in treatment or cure strategies where a durable prevention of viral rebound might be needed.
Constantinos Petrovas (Abstr. 167,
webcast), another presenter from the NIH Vaccine Research Center group, unveiled bispecific antibodies as another tool to clear persistent HIV out of hard-to-reach reservoirs. Follicular helper CD4 T cells are located within the germinal centers of lymph nodes and represent a major contributor to the latent reservoir. Bispecific antibodies that target HIV Env and CD3 are being developed to eliminate the latent reservoir by activating HIV from CD4 T cells and inducing killing of those cells by CD8 T cells. Phenotypic analysis of tonsillar cells revealed a memory population of follicular CD8 T cells. These follicular CD8 T cells exerted a superior killing capacity judged by in vitro mobilization of the effector molecules Granzyme B and perforin. Of all tissue CD8 T cell populations tested, these follicular CD8 T cells had the greatest ability to mediate killing of HIV-infected target cells after cross-linking with an anti-HIV Env/anti-CD3 bispecific antibody.
The field continues to move "towards a cure." Overall, these presentations at CROI offered an exciting and detailed new picture of persistent HIV infection in patients on ART. The picture requires further clarification. It appears to be more and more practical to achieve and maintain full suppression of HIV viremia in more and more people across the globe. But further advances in therapy might achieve eradication of persistent HIV infection. This must remain a goal of careful, determined, and focused research.
|
| |
|
|
|
|
|