 |
|
|
| |
CROI 2015 Metabolic Report: Vital Organs and HIV (and commentary by Jules Levin)
|
| |
| |
David Alain Wohl, MD - The University of North Carolina at Chapel Hill
(Commentary by Jules Levin below following this report by Dr. Wohl)
Introduction
Let's get things straight from the start: I am an unapologetic metabolic complications of HIV skeptic. The whole residual virus -> immune activation -> inflammation -> Yikes! dogma that has prevailed since someone drew inflammatory markers on a successfully treated HIV-positive patient makes sense to me on paper yet, in practice, not so much.
It is not that I do not believe that some people living with HIV infection have simmering immune responses that can be deleterious over time. I do. But, I have been very hesitant, based on the available evidence, to accept the 'sky is falling' alarm that this retrovirus is rapidly clogging coronary arteries and crumbling bones. Granted, many of us have patients who have experienced a heart attack or an atraumatic fracture at a young age. However, are these individuals representative of a pervasive phenomenon or are they outliers on a spectrum?
I tend to favor the latter and reports of rates of MI and stroke in HIV-infected patients decreasing over time and the wide splay of dots on scatter plots associating markers of immune activation or inflammation and [place scary outcome here] bolster my conviction that what we have may be more an excess of concern rather than an excess of risk. If my patients were living in an inflammatory storm, why were so many weathering aging so well? Could it be that just like a subset of HIV+ people have huge increases in visceral fat on ART, there is similarly a phenotype of patients who have vigorous responses to HIV that could lead to organ damage? If so, then identifying this risk group is where research should be pointed.
Instead we have studies showing an association between HIV infection and inflammation and bad outcomes in general. Anyone who has come within 10 feet of me knows my position that the field is dogged by the issue of confounding - that is, the messiness of studying events that are concentrated in people who have many of the risk factors for the very event of interest. Separating the HIV wheat from the confounder chaff is not easy, or always possible.
So, it is with this somewhat jaded perspective that I headed to Seattle for CROI2015. There I, admit, I did find some of the most compelling data ever presented on the well-being of vital organs in people living with HIV. While I remain convinced that talk of heightened risk of cardiovascular disease and accelerate aging in the setting of HIV infection is exaggerated, heading home I was impressed by these findings and highlight them below.
Heart
Excess Risk
One of the larger studies to look at excess risk of fatal cardiovascular disease in those living with HIV infection was conducted using 2001-2012 mortality data of New Yorkers (Hanna D, et al . #729, poster/report). The New York City HIV Surveillance Registry was crossed with the city's Vital Statistics Registry and the National Death Index to determine if there were more cardiovascular events in those with HIV (spoiler alert: there was). With over 145,000 HIV-positive people included and more than 29,000 deaths, this is a powerful data trove. Over time, as HIV-infected persons were less likely to die of HIV-related causes, the proportion succumbing to cardiovascular disease increased. Not really a surprise, but important to understanding how changes in the natural history of HIV can shift mortality data.
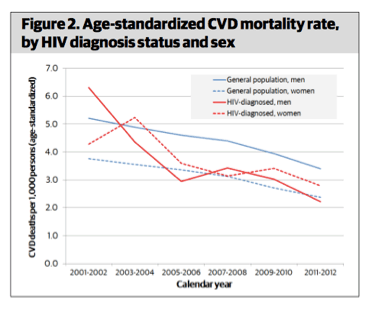
However, the cardiovascular disease mortality rate decreased in HIV-infected people, as well as in the general population over the study time period. Despite this universal downward trend, HIV infection was associated with a greater risk of dying of cardiovascular disease, especially for those younger than age 65 and individuals with an HIV RNA level that was over 400 copies/mL. Confounders like smoking and other major risk factors for heart disease and stroke were not able to be adjusted for and one has to believe that these play a role in the higher rates of CVD death in the HIV+ New Yorkers.
What this means for providers and patients: This study is instructive for a number of reasons. Foremost, like previous reports from the Kaiser Permanente group in California, it offers an undisputed truism: people with HIV infection do have higher rates of cardiovascular disease events (in this case, fatal events) compared to uninfected people. But, the risk of such outcomes is decreasing, not increasing, over time. Additionally, this and similar studies do not tell us is what drives this excess risk and to what extent this represents an excess of traditional risk factors versus HIV-related contributions via inflammation, medications, or both. It is certainly possible, if not probable, that the persons with HIV-infection who died in New York during this time period were disproportionately smokers, or cocaine users; many may have been at greater CDV risk for other traditional factors given who gets infected with HIV. Thus, while some will point to this study to shout out excess risk, this does nothing to forward an argument that HIV is the cause. The association of higher viral load levels and cardiovascular disease death is interesting and may be a signal of some virus-related influence, but this too is fraught with its own set of confounders.
Other studies looked at estimating CVD risk using a variety of methods including the Framingham Risk Score and the more au currant American College of Cardiology/American Heart Association (ACC/AHA) algorithm. In large cohorts of HIV+ patients, these calculators underestimated CVD risk (Thompson-Paul A, et al. #747, poster; Regan s, et al. #751, poster). These echo an early report from the D:A:D study suggesting that Framingham under-called risk in that cohort (Law MG, et al. HIV Med 2006).
What this means for providers and patients: The take home from these CROI analyses is that HIV infection may be a fudge-factor that has to be considered when assessing CVD risk. In my clinic, I make clear that such estimates are just that, estimates, based on populations. Unaccounted for factors related to living with HIV infection may increase the calculated risk of heart attack or stroke but whether that is the case for the patient in front of me no one can say.
Drivers of CVD Risk: Immune Activation/Inflammation
What accounts for this excess risk that remain unmeasured by these calculators? The finding of residual immune activation, particularly involving monocytes, in some studies of HIV+ patients with controlled viremia, has stoked intense concern that such an immune response could contribute to organ disease. Steve Grinspoon and his colleagues have elegantly postulated that activation of monocytes in HIV infection may be a root cause for excess risk of CVD (Subramian, et al. JAMA 2012) - including in a plenary at the conference (http://www.croiwebcasts.org/console/player/25786?mediaType=podiumVideo&). On-going activation of monocytes, in this model, leads to inflammation at the arterial wall, promoting atherosclerotic lesions. In HIV-uninfected patients, immune activation has been demostrated to play an important role in the genesis of the most dangerous types of coronary plaques and it is hypothesized that this is even more operative in the setting of heightened immune responses in HIV infection.
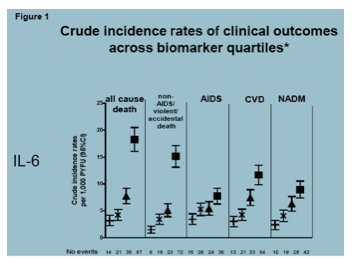
A number of studies looked to correlate markers of immune activation and inflammation with outcomes including death and CVD. One of the first studies to alert us to a role of inflammation in the well-being of people with HIV infection was the SMART trial, where baseline levels of CRP and D-dimer were associate with the odds of subsequent mortality. In an updated analysis, data from the ESPIRT trial were added and these markers as well as IL-6, another surrogate for inflammation, examined (Borges A, et al. #761, poster). Here, IL-6 was found to be a stronger predictor of all cause mortality and many fatal non-AIDS events than the other two markers. Adjustement attenuated the associations but IL-6 remained significant including for CVD.
Correlation between IL-6 and carotid intimal thickness (CIMT) was looked at in 149 patients in the US based SCOPE cohort (Hsu D, et al. #752, poster). Participants were almost all male, two thirds were white, a quarter were smokers and a third had diabetes. Median age was 48. Median CD4 cell count and all had suppressed HIV RNA levels. IL-6 was associated with CIMT after adjustment for age gender, race, CVD history, diabetes, hypertension, smoking, CD4, and LDL cholesterol and triglycerides. In addition, the monocyte was implicated here too with CCR5 expression also correlated with greater CIMT. Other inflammation or monocyte activation markers were not significantly linked to carotid thickness.
Another study of CIMT and monocyte activation looked at changes in CIMT over time in 50 HIV+ patients that demographically looked similar to the SCOPE participants described above, albeit fewer smokers and diabetics (Chow D, et al. #754. poster). In this study, CIMT was associated with, so called, non-classical monocytes and the monocyte activation marker MCP-1. Here, IL-6 was not reported to be associated with CIMT.
What this means for providers and patients: These were just of a couple of a number of studies presented at CROI and dozens of others that have been published demonstrating heightened levels of markers of inflammation in people living with HIV and a correlation of theses to adverse outcomes or their surrogates. Collectively, these investigations are a smoking gun, implicating immune activation, at least, as a part of the picture of excess risk for CVD and other end-organ disease in HIV. They are complicated and they do not always identify the same marker as being significant - likely reflecting differences in methods and populations studied. Moreover, we are still left to wonder the extent of the role of immune activation and inflammation as a causative agent for disease, rather than contributor or byproduct of other factors that may be more common in those who are HIV+. Few studies include HIV-uninfected persons and those that do are challenged to identify controls with risk factor profiles approximating those with HIV. So immune activation and inflammation happens. No doubt. How much does it drive end-organ disease and what can we do about it are what remain to be better understood.
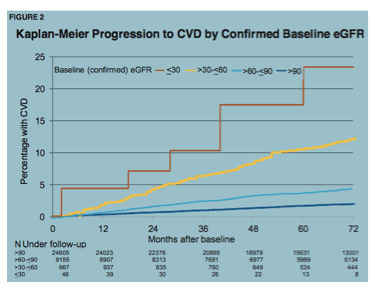
Drivers of CVD Risk: Renal Impairment
There was a slew of studies looking to associate a marker of something with cardiovascular disease or surrogates, thereof. One of the most important of these came from the D:A:D study group, which examined the association between CVD and a very common but under-appreciated risk: chronic renal disease (Ryom L, et al. #742, poster). Among almost 35,000 participants with at least two estimated GFR measurements, and available CD4 cell count and HIV RNA data, there were 1,251 CVD events experienced by 1,033 patients. The risk of a CVD event was strongly predicted by poor renal function (eGFR <30 ml/min/1.73m2). One in four of those with the lowest eGFR at five years of follow-up would be expected to have a CVD event. After adjustment for a number of reasonable confounding factors, there was a trend toward excess CVD events among those with middling eGFR 30-60 ml/min/1.73m2.
What this means for providers and patients: As described above, there is much talk in 'inflamo' circles of elevated lipids, inflammatory markers, coagulation factors and such, yet, these results highlight the lack of attention that has been paid to renal function when considering risk for CVD and cardio-protective interventions for those who are HIV-infected. As below, the channeling of patients with renal issues from tenofovir to abacavir muddies analyses looking at associations between this agent and CVD. Clinically, we must appreciate that renal insufficiency places our patients at relatively high risk of CVD and risk reduction interventions should be optimized in these individuals.
Statins do the Most Amazing Things
The CVD studies that garnered most attention at CROI were those that looked at interventions, especially statins. These ubiquitous agents, in addition to their desirable lipid-lowering effects, have purported anti-inflammatory properties and for those focused on unbridled inflammatory reactions triggered by HIV, they are truly irresistible.
In the SATURN-HIV Trial, the effect of rosuvastatin versus placebo on CIMT and the presence coronary plaque and coronary calcium score was studied in a select population of 148 HIV+ patients on ART (mostly men, non-white, 60% smokers, median CD4 ~600/mm3, and >75% undetectable HIV RNA) with a LDL-cholesterol of less than 130 mg/dL and elevated markers of immune activation or inflammation (Longenecker T, et al. #137, webcast). The idea was to replicate, in a smaller fashion, the JUPITER Trial of statin therapy in HIV-uninfected patients with elevated CRP (get it? Saturn, Jupiter).
Statin therapy appeared to arrest progression of CIMT as the active treatment arm saw little change in this parameter at 96 weeks while the control arm experienced an increase. The effects on coronary calcium and plaque were mixed. Among those with coronary calcium at baseline, statin therapy halted progression in its tracks compared to an increase in calcium scores in the controls. However, there was no difference in the prevalence of plaques or in calcified plaques in the study arms at week 96. Higher baseline monocyte activation markers and IL-6 levels were associated with a greater decline in CIMT.
A smaller, shorter-term randomized trial looked at whether atorvastatin could reduce coronary plaque progression in patients with at least a single plaque at baseline (Lo J, et al. #136, webcast). Here, 40 patients were randomized and at a year, plaque volume decreased 4.7% with atorvastatin but increased 18.0% in the placebo arm. There was a reduction in high-risk morphology plaques by 19% in the statin arm and a 20% increase among those receiving placebo.
What this means for providers and patients: These are very impressive results for statins and rival anything seen in studies of the uninfected. It remains unclear what the statins are doing but there does seem to be more going on than simply LDL-lowering. It should be appreciated that the most robust results, as far as atherosclerosis is concerned, were seen in those with evident abnormalities at baseline and it may be that these are individuals of an immune activation-inflammation phenotype that is most responsive to such an intervention.
The US AIDS Clinical Trials Group (ACTG) is launching a 6,500 person placebo controlled, randomized trial of statin therapy in those at lower risk for CVD events to determine if this therapy reduces actual outcomes like MI, stroke, and death. Called REPRIEVE, this study will answer many questions about the benefits (and disadvantages) of statins in HIV+ persons (http://reprievetrial.org).
More Abacavir and CVD
I am getting too old to be writing again about whether abacavir does or does not cause coronary artery spontaneous combustion. That this debate endures says as much about the difficulty of determining cause-and-effect as it does the limitations of observational cohort studies - the battleground on which this argument has been waged. It had been hoped that data from the NA-ACCORD, a large collection of longitudinal studies of HIV+ persons in North America, would cast as close to a deciding vote as there ever would be. Not at this CROI.
The results as presented were too wishy-washy to move partisans from their current viewpoints and too vague to convince the undecided (Palella F, et al. #749LB, poster). Although there are strengths of this study, including excellent ascertainment of outcomes, the results were presented in a way that aimed to be complete but instead led to collective head-scratching.
Analyses were conducted on two populations: a Full population (n=16,733) that included all ART users EXCEPT those on abacavir at cohort entry, and a Restricted population (n=6,485) of only those who were ART-naive on entry and who initiated ART some time thereafter (this was done to look at those with a clear start date of abacavir). There were 301 MIs in the Full and 93 MIs in the Restricted populations.
In the Full population 1,948 subsequently initiated abacavir and in the Restricted population, 486 took abacavir at some point. Interestingly, abacavir initiators were more likely to be black, IDU, have heterosexual risk, HCV infection, hypertension, renal impairment (see above), high total cholesterol, a CD4 <200/mm3, and a history of AIDS (i.e., confounding was a-bounding). Recent abacavir use was associated with MI in the Restricted population but not the Full population. The association between abavavir and MI in analyses that replicated the D:A:D analyses diminished with greater adjustment for traditional risks and comorbid conditions.
What this means for providers and patients: Heck, if I know. Mostly, it can be said that we still see here a signal linking abacavir and MI but that with the adjustment of confounders the association weakens. A major finding, again, is the very strong association between low eGFR and MI. That people with renal insufficiency are channeled toward abacavir makes it hard to tease this all apart. As more data are fed into this study there will be greater clarity but probably never to a degree sufficient for those who have already made up their minds.
Kidney
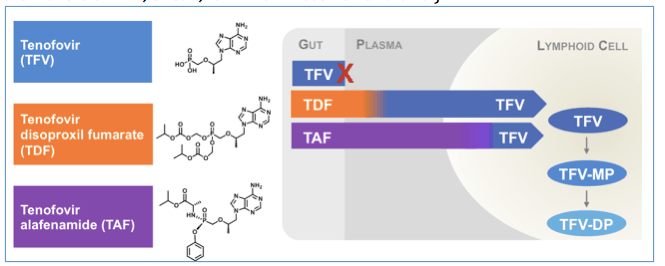
TAF: Like TDF but, less (protein) spilling
In addition to the D:A:D association between renal insufficiency and CVD, the most impressive kidney-related results presented at CROI were from two reports detailing the effects of tenofovir alafenamide, abbreviated as TAF, on renal parameters. Like tenofovir disoproxil fumarate (TDF; Viread), TAF is a pro-drug of tenofovir (TFV). However, rather than being converting to TFV predominantly in the plasma, TAF is metabolized to TFV once within the cell. This leads to a substantial reduction in circulating TFV levels, which are hypothesized to be associated with the bone and renal toxicity of this agent. In early studies, TAF had much less impact on renal function and bone density than TDF. (Full disclosure: I participated as an investigator in the TAF studies presented and serve on advisory boards convened by the makers of TAF, Gilead, for which I receive honoraria.)
The combined results of two large randomized trials comparing elvitegarvir/cobicistat/emtricitabine/TDF (E/C/F/TDF; Stribild) versus elvitegarvir/cobicistat/emtricitabine/TAF (E/C/F/TAF) in over 1,700 treatment naïve patients were presented (Sax P, et al #143LB, webcast, and Wohl D, et al. #113LB, webcast). Both regimens were found to be efficacious with over 90% achieving virologic success at week 48, and they were well tolerated. During the trial there were no patients who met criteria for tubulopathy in either study arm. In the E/C/F/TDF arm, four participants experienced renal events that led to treatment discontinuation, while no patients stopped E/C/F/TAF for renal toxicity. Over 48 weeks declines in eGFR were seen in both study arms, as expected, given all were receiving cobicistat, an agent that blocks creatinine transport from the blood to the urine; however, there was a slightly smaller drop among those receiving TAF. Urinary markers of renal tubular function including protein and albumin to urine creatinine ratio, retinol binding protein, and beta-2-microglobulin (B2M) increased in those on TDF but, TAF had marginal impact on these parameters, except for B2M, which declined substantially. As described below, bone density was remarkably better with TAF than TDF. Lipids were better with TDF and that likely reflects the lipid-lowering effects of circulating TFV.
A separate study examined the effect of switching to E/C/F/TAF from other regimens in patients with suppressed viremia but impaired renal function with eGFR between 30 to 69 ml/min/1.73m2 (Pozniak A, et al. #795, poster).
This cohort of 242 patients was older than that in the treatment naïve studies, with a median age of close to 60 years; 40% had hypertension, and 14% diabetes mellitus. Two thirds were on a TDF-containing regimen prior to study entry and 22% were on abacavir. Over 40% were also taking a boosted protease inhibitor. Switching preserved viral suppression in 92% with only three virologic failures.
There were no cases of tubulopathy and only two patients quit the study drug due to renal events: both likely related to hypertension. There was no significant change in eGFR after the switch to E/C/F/TAF whether calculated by Cockcroft-Gault or by use of cystatin C, a marker that, unlike creatinine, is not affected by cobicistat. In addition, rates of proteinuria and albuminuria improved and spilling of retinol binding protein and B2M into the urine also diminished with the change to the TAF regimen. In a substudy of about 75 patients who underwent measurement of actual GFR using iohexal clearance there was no change from baseline to week 24 after the switch.
What this means for providers and patients: The large phase III studies and the switch study impressively demonstrate a high degree of renal safety of TAF compared to TDF and serves notice that this big nuke is coming. It surprises me how many clinicians remain unaware of TAF even though E/C/F/TAF is expected to be available by the end of 2015 and next year we can expect a TAF/FTC tablet. Most of these providers will be using this new and improved Truvada or see it combined in new single tablet formulations like with rilpivirine or with darunavir/cobicistat. Just wait.
Body Shape
Protease inhibitors not a direct cause of big bellies
One of the most misunderstood metabolic complications of HIV therapy has been body shape changes. The early experience of fat accumulation within the belly and skinny arms, legs and face have been indelible and there continues to be a hard to shake belief that visceral fat accumulation is a protease inhibitor specific adverse effect. In reality, it is not and several carefully conducted studies show that lipohypertrophy can occur with other classes of ART. Confusing the issue is a general increase in generalized body fat after any successful HIV regimen is started - especially in those with lower CD4 cell counts.
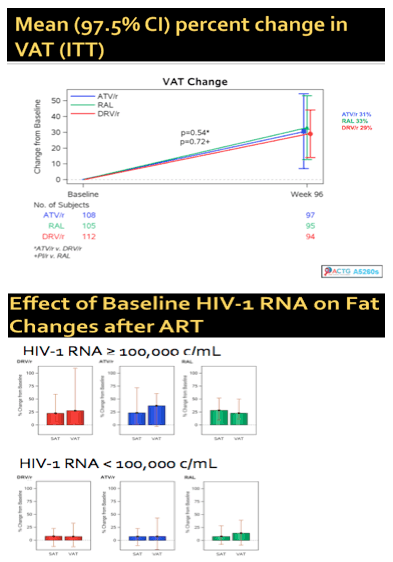
A substudy of the large three-arm ACTG trial comparing TDF/FTC plus raltegravir or darunavir+ritonavir or atazanavir+ritonavir (ACTG 5257) looked at changes in body shape after initiation of these regimens (McComsey G, et al. #140, webcast). Over 96 weeks, visceral fat as measured by CT scanning increased similarly in the protease inhibitor and the integrase inhibitor treated patients and there was no difference in the change in fat by study arm. Limb fat also increased in all study arms similarly. Interestingly, baseline HIV RNA levels were strongly correlated with changes in fat such that those with higher viral loads experienced greater increases in fat gain.
What this means for providers and patients: These results should hopefully bust the protease paunch myth. Modern ART, regardless of drug class, is associated with generalized increases in fat. Here protease inhibitor- and integrase inhibitor-based ART led to essentially identical changes in body shape. This suggest that these changes are less an antiretroviral effect than a result of effective treatment of HIV. Those with higher HIV RNA had greater changes in fat. The reason for this is unclear. Perhaps those with high levels viremia have more advanced disease. Data on associations between baseline CD4 cell count or body mass index were not presented. Overall, patients and providers should not shy away from protease inhibitors due any fear of belly fat.
Bone
TAF effects on bone
In the large naïve trials of E/C/F/TAF versus E/C/F/TDF, bone density was also measured by DEXA scans of the hip and spine. This is the largest study of bone density in HIV+ people ever with almost 1,700 patients assessed at three time points over a year (Sax P, et al #143LB, webcast).
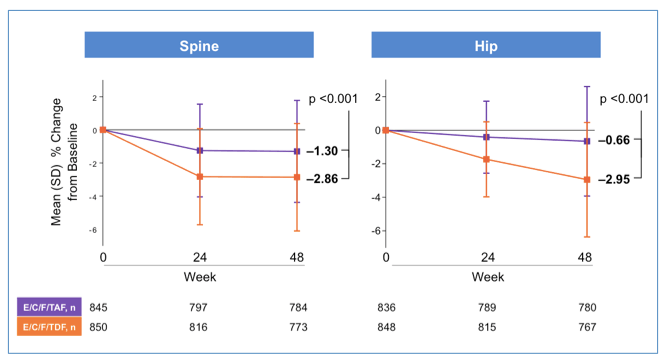
TAF was shown to have much less impact on bone density than TDF and at the hip the change from baseline was fairly negligible. About 70% of those on TAF had no clinically significant change in bone density during the first year of therapy, while half of those on TDF saw a significant decline.
What this means for providers and patients: We have tolerated the effects of TDF on bone, largely because of its other attributes, and because we often do not measure bone density clinically. But, bone issues is likely to become more of a concern for HIV providers as their patients age. Therefore, the findings or a minimal impact of TAF on bone density is welcome and will certainly help justify use of this agent in place of its antecedent.
Statin effects on bone
The magic of statins to make the world a better place for people living with HIV was put to the test in the SATURN-HIV trial (see above). However, while there was a positive impact seen in terms of carotid intimal thickness and in progression of coronary plaques, rosuvastatin was not found to do much to change bone density and may worsen insulin resistance (Erlandson K, et al. #771, poster). DEXA scans done at baseline and weeks 48 and 96 after start of the statin found no significant difference in bone density at either the hip or spine between the first and last time point compared to placebo. Estimation of insulin resistance was found to be worse with rosuvastatin than placebo - a worrisome finding given an association between statin therapy and diabetes in studies of HIV-uninfected patients.
What this means for providers and patients: Statins should be used as indicated for the prevention of CVD. There is increasing interest in applying statin therapy to reduce inflammation and its sequalae in people living with HIV infection. While the above mentioned results from small studies looking at carotid intimal thickness and coronary plaque show promise, it is premature to use these agents broadly to treat HIV-associated inflammation. The results of the SATURN-HIV Trial make clear that rosuvastatin's effects did not translate into improvements in bone density, although such an effect has been seen in the HIV-uninfected. Reasons for this difference are not clear and may point to very different mechanisms of bone loss based on HIV status. That there may be a cost vis-a-vis glycemic control lends a cautionary note to the enthusiastic tunes many are singing about these agents. Fuller study of the good and not so good aspects of statins in HIV+ patients are needed and the REPRIEVE trial will certainly be valuable in addressing these important questions.
Assessing bone density
As is the case for CVD risk assessment, estimation of the odds of a bone fracture can be a challenge. DEXA scanning can provide data on bone density and this can be correlated to fracture risk, but the test is not always readily available, incurs some radiation exposure, and may not be covered by health insurance. The FRAX is a web-based calculator created by the WHO that acts much like the Framingham Risk Score for 10-year bone fracture risk. How well FRAX works in predicting fractures in HIV+ persons has been debated.
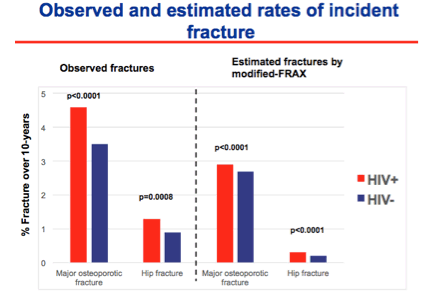
The ability of FRAX to predict fractures was studied in the US Veterans Aging Cohort Study's Virtual Cohort (Yin M, et al. #141, webcast). Data from men 50-70 years of age, including over 7,000 HIV+ men and over 17,000 HIV- men, were used to calculate a FRAX score and this was correlated to ICD-9 coding of actual fractures.
Overall, HIV+ men had higher rates of fractures than HIV- men and this is not unexpected given more traditional fracture risks among the infected men (lower weight, more prior fractures, more alcohol and corticosteroid use). Importantly, FRAX underestimated fracture risk in both HIV+ and HIV, but was worse in predicting risk in the HIV+. In fact, when applying the results to thresholds for use of pharmacological intervention, 97% of HIV+ men with an actual fracture would have not been flagged for treatment based on their FRAX score. Adding HIV infection as a 'secondary' cause of osteoporosis increased the accuracy of the score somewhat.
What this means for providers and patients: The FRAX has been a convenient and cheap method to assess fracture risk but the poor showing of the calculator in this study makes it hard to rely on it henceforth. The problem is that there are not great alternatives. DEXA scans are not as accessible or affordable and they too may not be as accurate as one would desire. At this point, a DEXA in those at risk (age 50+) remains preferred. When DEXA is not available the FRAX should be interpreted with some caution and can be assumed to underestimate risk substantially in men such as those studied here.
--------------------------
Comments From Jules:
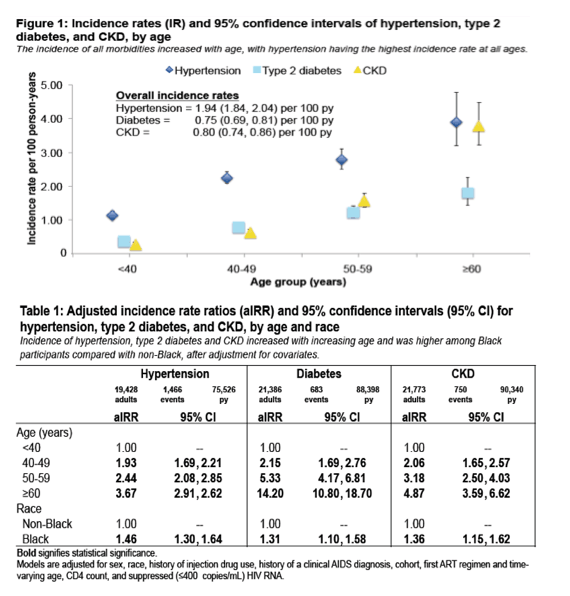
I think inflammation & activation due to HIV is occurring & does contribute to the increased & earlier development of comorbidities. Behavior & lifestyle can contribute as well, including IDU & substance abuse history. There are too many studies implicating HIV and its associated inflammation/immune activation for this not to be a causative factor towards increased and earlier onset of comorbidities. Several studies find onset of immunosenescence soon after HIV infection, in adults and a study at CROI 2015 in children, and studies associated immunosenescence with heart disease in HIV. To underscore this a study at CROI (Utay et al. #47, webcast/report) found very early ART, median time since acquisition to HIV was 16 days, improved most markers of microbial translocation, inflammation, coagulation and fibrosis, but ultimately suppressive ART started during acute HIV infection does not eradicate the inflammation associated with increased morbidity and mortality in treated chronic HIV infection, thus suggesting that since ART decreased the biomarkers that HIV does cause these biomarkers to increase. And a 2nd study finds similarly (Crowell et al. #48, webcast/report) - detectable HIV RNA is common during AHI (acute HIV infection) and tends to occur during the earliest stages of AHI, viral infiltration is broad and rapid - detectable HIV RNA during AHI correlates with: increased HIV burden in the blood, colon and CSF, systemic inflammation, depletion of colonic CD4 cells, CD 8 T-cell activation in the blood and colon - and after 24 weeks of ART, differences in viral burden, inflammation and T-cell activation largely disappear; again suggesting that HIV is a major culprit since ART reduces biomarkers. I have reviewed a few unique select studies in this commentary. Comorbidities themselves can cause inflammation. But residual HIV in HIV+ with undetectable HIV causes inflammation/activation, and immune exhaustion & senescence which all contributes or drives accelerated aging and higher risk for comorbidities at earlier ages; and as a person ages over 50-60 years old, when the immune system declines in HIV- and HIV+ this results in an increasingly greater decline in HIV+. I agree this may not affect all patients the same way. We are seeing an increase in comorbid conditions particularly increasing as HIV+ age. In this study at CROI the objective was to estimate the incidence of hypertension, diabetes and chronic kidney disease in a large collaboration of HIV cohorts (NA-ACCORD) (Wong et al. #1053. poster). They found older adults and Black adults had the highest incidence of the three comorbidities looked at in the study. As you can see in Figure 1 below the incidence rates increase quite a lot from <40 years of age through over age 60.
In this study at CROI 2014 (Greene et al. #766, poster) they found more than half of participants had 2 or more geriatric syndromes, which was associated with CD4 nadir, 86% of subjects had at least one geriatric syndrome and 54% had 2 or more syndromes, and (9%) subjects met the full criteria for frailty, while 79 (56%) met criteria for pre-frailty.
What about microbial translocation? There have been numerous studies linking microbial translocation in HIV+ to immune activation and inflammation. In this study at CROI (Vera et al. #477, poster). the authors say "microbial translocation is characterized by the translocation of microbial products derived from the gastrointestinal tract of HIV-infected individuals to the bloodstream". HIV damages the gut. The aim of this study was to examine the relationship between a marker of microbial translocation (ribosomal 16s DNA) and brain biomarkers of inflammation, function and structure in treated HIV-infected individuals. They found in neuroasymptomatic treated HIV-infected individuals microbial translocation is associated with markers of neuroinflammationand abnormalities in white matter integrity.
At CROI 2015 Hadigan et al. (#756, poster) found HIV-infected subjects demonstrate subclinical impairment in systolic function and this is associated with markers of chronic immune activation, inflammation and tissue remodeling.
Studies in adults & children found senescence in HIV+ at CROI. In "Chronic HIV Infection Exacerbates Cellular Aging Markers in Isolated T-Cell Subsets" (Hsieh et al. #306, poster), the study aim was to investigate the possible role of HIV and/or cART on immune aging. HIV+ ages 6-70 were studied, and the authors report "these results suggest a potential relationship between HIV infection and shorter TL (telomerase) in proliferative CD8+ T cells suggesting a role in HIV immune cell aging. In contrast, cART duration was related to mtDNA oxidative damage in CD8+ T cells, suggesting that cumulative exposure may play a role in mitochondrial aging in this immune compartment", and HIV causing immunosenescence.
Increased cardiovascular (CV) risk persists among patients with treated HIV disease, and chronic immune activation is thought to contribute to this excess risk. Carotid intima-media thickness (CIMT) assesses atherosclerotic burden and predicts future CV events. At CROI 2015 Hsu et al (# 752, poster) studied the immunologic correlates of CIMT in patients on ART with suppressed viral load (VL), and found In patients on ART with suppressed VL, higher plasma IL6 and CCR5 expression on monocytes and higher % of CD57+ cells in CD28-CD8+T cells were independently associated with thicker CIMT after adjusting for CVD risk factors and CD4 count. Dysfunction of innate immune cells and CD8 T cell senescence likely contribute to atherosclerosis in the setting of treated and suppressed HIV. There is growing evidence that the CD8+CD28- (CD8+CD57+) T-cell population plays a significant role in various diseases or conditions, associated with chronic immune activation such as cancer, chronic intracellular infections, chronic alcoholism, some chronic pulmonary diseases, autoimmune diseases, allogeneic transplantation, as well as has a great influence on age-related changes in the immune system status (Strioga, Immunology. 2011 Sep; 134(1): 17-32).
At CROI - "Premature Aging and Immune Senescence in HIV-1-Infected Children" (Gianesin et al. #923, poster) - in HIV+ children perinatally infected aged 0-5 the proportion of CD8+ cells with senescent phenotype (CD28-CD57+) was higher compared to HIV exposed-uninfected and unexposed-uninfected. HIV-1-infected children had a lower telomere length compared to age- and gender- matched controls, suggesting an accelerated biological aging; - age-adjusted telomere length is shorter in ART-untreated than in ART-treated children, thus suggesting that HIV-1 itself, rather than exposure to antiretroviral drugs, influences the senescence process. ART does not restore full functionality of the immune system, and health status remains characterized by a number of non-AIDS defining complications associated with aging, including malignancies, even among long-term ART-treated patients. Therefore, HIV-1-infected subjects may suffer of premature and accelerated aging; chronic immune activation, due to persistence of HIV-1 virions may play a key role in this senescent pathway.
Again in children (Purdy et al. #928, poster/report): this study was to measure biomarkers of cardiovascular injury and immune activation in relationship to coronary plaque burden in patients infected with HIV early in life in HIV+ and HIV-negative controls: 5 adolescents & young adults who acquired HIV early in life and 11 healthy controls.
They find: individuals infected with HIV early in life have higher levels of immune activation, inflammation, and circulating adhesion molecules than controls; viral suppression of HIV helps reduce but not completely reverse the chronic inflammatory state of HIV; Immune activation, in particular %CD8+CD38+DR+, is associated with coronary plaque in this cohort of patients with HIV.
HIV-1 induces a strong immune activation, which is particularly evident within the CD8+ T-cell compartment. The elevated and chronic stimulation induced by HIV-1 may result in the exhaustion of the capacity to generate new T-cells. These characteristics are not unique to HIV infection, but they are also common to other conditions that result in some degree of immunodeficiency, like ataxia telangiectasia and normal human ageing. They may reflect a premature decline of the immune resources necessary for viral control and therefore contribute to the onset of disease progression. Our data indicate that HIV-1 infection results in immune activation not only directly, but also indirectly, with the activation of cells specific for non-HIV antigens . The level of CD8+ CD38+ T lymphocytes in blood correlates with disease progression in HIV-infected individuals, independently of the CD4 count. Effective antiretroviral therapy reduces this lymphocyte subset in parallel with plasma viremia, although CD38 expression on CD8+ cells does not normalize completely in most subjects, and might reflect residual HIV replication. HIV disease progression correlates with increased proportions of highly differentiated CD8+ T-cells, which exhibit characteristics of replicative senescence and probably indicate a decline in T-cell competence of the infected person. The differentiation of CD8+ and CD4+ T-cells towards a state of replicative senescence is a natural process. It can be driven by excessive levels of immune stimulation. This may be part of the mechanism through which HIV-1-mediated immune activation exhausts the capacity of the immune system. (PLoS Biol 2(2): e20; 2004, Appay et al).
Since HIV disease involves accelerated immunological aging [3], research that focuses on a cell population that undergoes significant changes during chronological aging may provide novel insights into HIV pathogenesis. Indeed, both HIV disease and aging involve the accumulation of a population of dysfunctional CD8+ T cells with characteristics of cellular (replicative) senescence, an end stage characterized by irreversible cell cycle arrest, multiple genetic and functional changes, and shortened telomeres. The clinical relevance of this cell population is underscored by several observations. First, CD8+ T cells with several signature features of replicative senescence, including reduced anti-HIV effector functions, permanent suppression of CD28 gene expression, critically short telomeres, and loss of the ability to upregulate telomerase, are significantly increased in HIV-1-infected persons, even in those on ART [4]. Moreover, the abundance of CD8+CD28- T cells early in the infection is actually predictive of the subsequent rate of progression to AIDS [5]. Given the prolonged survival and "graying" of the HIV-infected population, it is possible that the CD8+ T cell defects due to the infection may synergize with similar defects associated with aging, further highlighting the critical need for more precise characterization of this subset. [PLoS ONE 8(5): e64702, Effros et al].
---------------------------------
|
| |
|
|
|
|
|