 |
|
|
| |
Grazoprevir/Elbasvir plus Ribavirin For Chronic HCV Genotype-1 Infection After Failure of Combination Therapy Containing a Direct-Acting Antiviral Agent
|
| |
| |
Download the PDF here
Jnl of Hepatology
April 2015
Xavier Forns, Stuart C. Gordon, Eli Zuckerman, Eric Lawitz, Jose L. Calleja,
Harald Hofer, Christopher Gilbert, John Palcza, Anita Y.M. Howe, Mark J.
DiNubile, Michael N. Robertson, Janice Wahl, Eliav Barr, Maria Buti
Abstract
Background & Aims
The Phase-2 C-SALVAGE study evaluated an investigational interferon-free combination of grazoprevir (a NS3/4A protease inhibitor) and elbasvir (a NS5A inhibitor) with ribavirin for patients with chronic HCV genotype-1 infection who had failed licensed DAA-containing therapy.
Methods
C-SALVAGE was an open-label study of grazoprevir 100 mg/elbasvir 50 mg QD with weight-based ribavirin BID for 12 weeks in cirrhotic and non-cirrhotic patients with chronic HCV genotype-1 infection who had not attained SVR after ≥4 weeks of peginterferon and ribavirin plus either boceprevir, telaprevir, or simeprevir. Exclusion criteria included decompensated liver disease, hepatocellular carcinoma, and HIV or HBV co-infection. The primary efficacy outcome was SVR12 defined as a HCV RNA level below the assay limit of quantification 12 weeks after the end of treatment.
Results
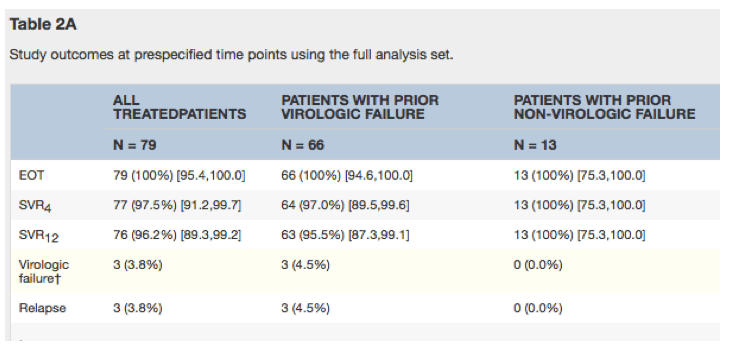
Of the 79 patients treated with ≥1 dose of study drug, 66 (84%) patients had a history of virologic failure on a regimen containing a NS3/4A protease inhibitor; 12 of the other 13 patients discontinued prior treatment because of adverse experiences. At entry, 34 (43.6%) of 78 evaluable patients harbored NS3 RAVs. SVR12 rates were 76/79 (96.2%) overall, including 28/30 (93.3%) patients with genotype 1a infection, 63/66 (95.5%) patients with prior virologic failure, 43/43 (100%) patients without baseline RAVs, 31/34 (91.2%) patients with baseline NS3 RAVs, 6/8 (75.0%) patients with baseline NS5A RAVs, 4/6 (66.7%) patients with both baseline NS3 and RAVs, and 32/34 (94.1%) cirrhotic patients. None of the 5 reported serious adverse events were considered drug-related.
Conclusions
Grazoprevir/elbasvir plus ribavirin for 12 weeks provides a promising new treatment option for patients after failure of triple therapy containing an earlier-generation protease inhibitor.
Introduction
The introduction of direct-acting antiviral agents (DAAs) set a new standard for HCV care, substantially increasing achievable rates of sustained virologic response (SVR) [[1], [2], [3]]. Although undoubtedly a quantum advance, a sizeable minority of patients treated with first-generation protease inhibitors combined with peginterferon alfa and ribavirin (PR) do not clear their infection. Virologic failure after DAA therapy is often accompanied by the emergence of resistance-associated variants (RAVs) which can limit subsequent treatment options [[4], [5], [6], [7]]. The signature NS3 RAVs for first-generation protease inhibitors have been well characterized in vitro, but their full therapeutic implications remain incompletely understood [[8], [9], [10]]. In particular, the extent and significance of in-class cross-resistance between first and later generation protease inhibitors have not been definitively established in the clinic [[10], [11], [12]].
Whether patients who have not been cured by triple therapy with PR and an older protease inhibitor can be reliably salvaged with regimens incorporating a more potent protease inhibitor with a higher genetic barrier to resistance together with a DAA of another class has not been comprehensively evaluated. Earlier studies with simeprevir plus sofusbuvir indicate that SVR12 rates exceeding 80% might be attainable in genotype 1 infection after failure of PR plus a first-generation protease-inhibitor [13]. Additional effective, well-tolerated, and convenient treatment options need to be identified for patients who are not cured by DAA ± PR combination regimens [[1], [3], [13], [14], [15], [16], [17], [18]].
The C-SALVAGE study investigated the safety and efficacy of an investigational combination of grazoprevir (a NS3/4A protease inhibitor) and elbasvir (a NS5A inhibitor) with ribavirin for patients with chronic HCV genotype-1 infection who had failed licensed DAA-containing regimens. Many RAVs selected by earlier protease inhibitors remain susceptible to grazoprevir [19]. The main objective of this phase 2 trial was to explore the utility of a novel interferon-free DAA-combination in patients who had not achieved SVR after triple therapy containing a DAA in the context of emergent RAVs. Specifically, C-SALVAGE was designed to test whether a DAA-regimen anchored by a non-cross resistant protease inhibitor could consistently clear HCV infection among patients with a history of failure on a triple regimen containing PR and a less active first-generation protease inhibitor.
Discussion
In the open-label C-SALVAGE trial, 79 patients infected with HCV genotype 1 who had failed combination therapy with PR and a licensed protease inhibitor were treated with grazoprevir/elbasvir plus ribavirin, including 43% with cirrhosis and 84% with prior virologic failure. HCV RNA levels were below the assay limit of detection in all patients at the end of treatment despite a high prevalence of NS3 RAVs at baseline. Relapses occurred in 3.8% during the first 12 weeks of follow-up, resulting in an overall SVR12 rate of 96.2%. SVR12 was achieved in 63 (95.5%) of the 66 patients with a history of virologic failure and in all (100%) of the other 13 patients with non-virologic reasons for failing earlier treatment. SVR12 was attained in 29 (90.6%) of the 32 patients with a history of past virologic failure harboring virus with documented NS3 RAVs conferring decreased susceptibility to boceprevir, telaprevir, and/or simeprevir at baseline. SVR12 rates in cirrhotics vs. non-cirrhotics and by infecting sub-genotype (1a vs. 1b) were similar.
The emergence of class resistance among drugs sharing a similar mechanism of action has long been a concern after failed treatment of viral infections. Fortunately with antiretroviral therapy, different drugs in the protease inhibitor class can often be effectively used in salvage combinations after failure of a first protease inhibitor. C-SALVAGE demonstrated that HCV-infected patients with genotype 1 failing triple therapy with PR combined with an earlier-generation protease inhibitor can be successfully retreated with a protease inhibitor-anchored regimen, provided that the new protease inhibitor is substantially more active and not cross-resistant to the failed protease inhibitor.
Our results are consistent with the recently published Phase 2 SIRIUS trial using the nucleotide polymerase inhibitor sofosbuvir and the NS5A inhibitor ledipasvir in patients with genotype-1 infection and compensated cirrhosis who had failed protease inhibitor-based regimens [[23], [24]]. By switching to 2 new drug classes, SVR12 was achieved in 96% with the 12-week regimen of sofosbuvir/ledipasvir plus ribavirin and in 97% with the 24-week regimen of sofosbuvir/ledipasvir without ribavirin. Both studies found high SVR12 rates in previous non-responders with compensated cirrhosis when treated for dual DAAs plus ribavirin for as short as 12 weeks. C-SALVAGE expanded the sofosbuvir/ledipasvir findings by showing that a potent, non-cross-resistant protease inhibitor like grazoprevir can be successfully used in patients failing earlier-generation drugs of the same class.
Another instructive observation from this trial is the high tolerability of the study regimen in patients who did not tolerate their earlier interferon-containing therapy. A completion rate of 78 (98.7%) among the 79 study participants (including 11 of the 12 patients who prematurely stopped their prior therapy due to drug intolerance) was accomplished despite the reuse of ribavirin, although ribavirin dose reduction was required in 11 (13.9%) patients.
The role of baseline resistance testing requires continued scrutiny as the use of different classes of directly-acting antiviral agents for chronic HCV infection becomes increasingly widespread [[2], [5], [6], [25]]. Not all baseline variants will actually confer clinically meaningful drug resistance [12]. Before interpretive guidelines for genotypic and phenotypic resistance testing can be established for a given drug, RAVs must be distinguished from therapeutically inconsequential polymorphisms based on extensive clinical correlation. Furthermore, specific RAVs may remain susceptible to other drugs within the same class. Because combination therapy is universally recommended, baseline variants might only negatively impact outcome when abundant RAVs, high-level resistance, cross-resistance to other co-administered directly-acting antiviral drugs, and/or erratic compliance with an unforgiving regimen are concurrently present. Further study is needed to fully define the impact of specific RAVs on the efficacy of grazoprevir/elbasvir in patients after failure of DAA-based regimens.
C-SALVAGE exclusively enrolled patients with failures incurred on the first 3 available protease inhibitors, so the utility of grazoprevir/elbasvir after failure of more recently approved directly-acting regimens cannot be assessed from these data. Likewise, it is not possible to independently evaluate the contribution of ribavirin to efficacy or toxicity because all patients received ribavirin. The need for ribavirin to boost efficacy in the setting of directly-acting antiviral regimens has varied with the specific drugs and circumstances [[2], [13], [14], [15], [16], [17]]. Recently published findings from the C-WORTHY program indicate that ribavirin is unlikely to play an essential role as an adjunct to grazoprevir/elbasvir [[26], [27]]. The phase 3 C-EDGE trials in treatment-experienced and other traditionally "hard-to-treat" populations given grazoprevir/elbasvir without ribavirin will help to better address these questions.
Our analysis has several potential constraints. The study was relatively small and open-label without a concurrent control group. Only patients unsuccessfully treated with earlier-generation protease inhibitor combined with PR were enrolled; despite the protocol inclusion criteria, no sofosbuvir-experienced patients were entered. Patients with hepatic failure were excluded. Cirrhosis was biopsy-proven in a minority of the cirrhotic patients enrolled in the trial; however, noninvasive assessments of liver fibrosis have increasingly replaced biopsy as the practice standard in this context. The reasons for failure on prior treatment regimens were heterogeneous, and included discontinuations due to intolerability as well as lack of efficacy. Signature NS3 RAVs were not detected at baseline in 33 (50.8%) of the 65 evaluable patients with a history of virologic failures involving early-generation protease inhibitors. By missing minor variants, population-based sequencing as used here likely underestimated the frequency of potentially relevant RAVs [20]. Since the limits of detectability can vary with sample, assay, and operating characteristics, patients with detectable but unquantifiable HCV RNA were regarded as successes [[28], [29]]; however, all patients with SVR12 had undetectable HCV RNA by a standard automated sensitive assay. Even though SVR12 has become the nearly universally accepted endpoint for HCV treatment trials (with endorsement from major regulatory agencies), the correlation between SVR12 and cure theoretically should be established for each individual regimen to exclude late relapses at week 24 and beyond [[28], [29], [30]].
In the C-SALVAGE trial, 79 patients with chronic HCV genotype-1 infections who had failed protease-inhibitor-based combination regimens were treated with grazoprevir/elbasvir and ribavirin, including 43% with cirrhosis and 84% with prior virologic failure. NS3 RAVs for first-generation protease-inhibitors were present in almost half of the patients at baseline but infrequently exhibited high-level cross-resistance to grazoprevir in vitro [19]. SVR12 was achieved in all but 3 patients with relapse. Therapy was generally well tolerated in this treatment-experienced population with only a single early discontinuation. The interferon-free regimen of grazoprevir/elbasvir plus ribavirin given orally for 12 weeks offers a promising new treatment option for patients who have failed therapy with PR and an earlier protease inhibitor [[31], [32]].
Patients And Methods
Study Design
C-SALVAGE was an international, open-label, hypothesis-generating study of grazoprevir (100 mg PO QD), elbasvir (50 mg PO QD), and ribavirin (given PO BID at a total daily dose of 800 mg to 1400 mg based on weight) for 12 weeks in patients with chronic HCV genotype-1 infection who had failed ≥4 weeks of peginterferon and ribavirin combined with boceprevir, telaprevir, simeprevir, or sofosbuvir. Adults ≥18 years of age with plasma HCV RNA levels ≥10,000 IU/mL at screening were eligible. Exclusion criteria included decompensated liver disease, hepatocellular carcinoma, HIV or HBV co-infection, thrombocytopenia <50 x 103/μL, or hypoalbuminemia <3.0 g/dL. Patients with compensated cirrhosis were not excluded but the proportion of cirrhotic patients in the study was limited to a maximum of 40%. To ensure sufficient numbers of enrolled patients with baseline NS3 RAVs, approximately 80% of the enrolled subjects were to have experienced virologic failure on prior triple therapy. Written informed consent was obtained from all participants. The trial was conducted in accord with Declaration of Helsinki and Good Clinical Practice guidelines. Subjects who discontinued treatment prior to completion were encouraged to return for all remaining study visits. Patients were to be followed for 24 weeks after the cessation of study therapy. The trial was initiated 23-May-2014 and will be ongoing until approximately 23-April-2015 when the last patient is scheduled to complete the final follow-up visit.
The protocol mandated staging of liver disease which could be accomplished by biopsy or noninvasive assessment within an appropriate timeframe. Cirrhosis was documented by a liver biopsy showing Metavir stage F4 at any time; transient elastography (Fibroscan) performed within 12 months of entry yielding a result >12.5 kPa; or biochemical markers of liver fibrosis (FibroTest or FibroSure) yielding a score of >0.75 coupled with an AST:platelet ratio index (APRI) of >2. The absence of cirrhosis could be inferred if a liver biopsy performed within the previous 24 months did not reveal cirrhosis, a Fibroscan performed within the previous 12 months had a result of ≤12.5 kPa; or a FibroSure or FibroTest score was ≤0.48 with an APRI of ≤1 in the preceding 12 months.
Viral and Resistance Assays
Plasma HCV-RNA levels were measured by the COBAS TaqMan v2.0 assay (Roche Diagnostics, Branchburg, NJ, USA) with lower limits of quantification and detection of 15 and 9 IU/mL, respectively. Specimens for viral load measurements were to be done at screening; baseline (Day 1); treatment weeks 1, 2, 4, 6, 8, 10, and 12; and follow-up weeks 4, 8, 12, and 24 after cessation of therapy. Specimens from all participants before initiation of study therapy were used to generate baseline HCV-subtype sequence information. Additional samples were collected from patients who met the criteria for virologic failure at the time of failure and at later follow-up visits. Due to assay limitations, only samples with HCV RNA titers ≥1000 IU/mL were sequenced.
NS3 and NS5A genes were amplified using reverse transcriptase-polymerase chain reaction (RT-PCR) followed by population sequencing with a lower limit of variant detection of approximately 20-25% prevalence [[19], [20], [21]]. Resultant amino-acid sequences were compared to wild-type HCV genotype 1a (H77) or 1b (Con1) reference sequences. Phenotypic characterization of variants was conducted using HCV replicons; resistance was characterized as low-level or high-level resistance based on the effective (inhibitory) concentration (EC50 ≤5x versus >5x of the wild-type referent strain, respectively).
To search for NS3 variants at baseline, all amino acid positions within NS3 protease were examined. Single NS3 amino acid substitutions involving V36A/G/L/M/I, T54A/C/G/S, V55A/I, Y56H, Q80K/R, V107I, 122A/G/R, I132V, R155X, A156S/T/V/F/G, V158I, D168X, I/V170A/F/T/V, and M175L were considered clinically relevant RAVs because these mutations had been commonly identified after treatment failures with boceprevir, telaprevir, simeprevir, or vaniprevir [12].
A subset of these first-generation protease-inhibitor RAVs (involving Y56H, R155G/T/W, A156G/T/V/L, and D168A/G/T/V/L/I/F/Y/E/H/K) exhibited a >5-fold increase in grazoprevir EC50 in genotype 1a replicons relative to the wild-type referent [19]. Post-baseline amino acid substitutions at loci 36, 54, 55, 80, 107, 122, 132, 155, 156 158, 168, 170, and 175 were used to define emergent RAVs in virologic failures on or after study therapy.
Statistical Analyses
Because C-SALVAGE was an estimation study without a control group, no formal hypothesis-testing was planned. The primary efficacy analysis prescribed by protocol estimated the proportion of patients without significant protocol violations (the per-protocol population) with a HCV RNA level below the limit of quantification (15 IU/mL) 12 weeks after the end of treatment (SVR12). Only observed success or failure contributed to the primary efficacy analysis. The 95% confidence intervals for SVR rates were computed by the Clopper-Pearson method [22].
The protocol-stipulated secondary efficacy analysis and the primary safety analysis were performed on all patients who received at least one dose of study treatment (the full analysis set). For this sensitivity analysis of efficacy, patients with missing outcome data were counted as failures unless flanked by visits where HCV-RNA levels were both <15 IU/mL. Adverse events occurring anytime during the treatment period and the initial 14 days of post-therapy follow-up were included in the safety analyses. Analyses based on the full data set form the focus of this report.
Exploratory analyses were performed for SVR4 and are planned for SVR24. Descriptive analyses were done for clinically relevant subgroups, such as patients with baseline RAVs and cirrhosis. SVR12 rates were computed by baseline NS3 RAVs categorized by their in vitro susceptibility to grazoprevir.
Results
Subject accounting and baseline characteristics
All 79 enrolled patients were treated with ≥1 dose of study drug (Fig. 1). There were 33 (42%) women, 2 (3%) non-whites, 34 (43%) cirrhotics (including 7 diagnosed by biopsy), and 30 (38%) genotype 1a infections (Table 1). All participants had received a NS3/4A protease inhibitor; none had taken sofosbuvir. From the dates provided in the medication summaries, we estimated that the median [interquartile range] time between prior and study therapy was approximately 72 [48,96] weeks. A total of 66 (84%) patients had a history of virologic failure. Of the remaining 13 patients with non-virologic failure, 12 had discontinued treatment because of drug intolerance or adverse events and 1 had received an abbreviated 12-week course of PR plus simeprevir as part of a clinical trial.
At entry, 34 (43.6%) of the 78 patients with available NS3 sequencing data harbored variants resistant to boceprevir, telaprevir, or simeprevir. Only 4 (11.8%) of these 34 patients with signature NS3 RAVs harbored variants with >5-fold decreased in vitro susceptibility to grazoprevir in a replicon assay. In addition, 8 (10.1%) of the 79 patients with available NS5A sequencing data harbored virus with NS5A polymorphisms at baseline, including 5/8 (62.5%) patients with variants exhibiting >5-fold decreased susceptibility to elbasvir in vitro.
A total of 78 (99%) patients completed therapy. Only 1 patient prematurely stopped treatment after 80 of the stipulated 84-day course due to dysphagia, dehydration, and vomiting attributed to radiation therapy being given for squamous cell carcinoma of the larynx and considered unrelated to study medications. This patient remained in the study for subsequent follow-up visits. One other patient who completed the prescribed course of study therapy dropped out of the study after relapsing at post-treatment week 4.
Virologic response
At the end of therapy, HCV RNA levels were <15 IU/mL in all 79 (100%) patients (Table 2A). Relapses occurred in 3 (3.8%) patients during the first 12 weeks of post-therapy follow-up (2 at follow-up week 4 and 1 at follow-up week 8). SVR12 was achieved in the other 76 (96.2%, 95% confidence interval [89.3,99.2]) patients, all of whom had undetectable HCV RNA at follow-up week 12. The 3 patients not achieving SVR12 had a past history of virologic failure. SVR12 rates were 63/66 (95.5%) in patients with prior virologic failure, 33/36 (91.7%) in patients with NS3 and/or NS5A variants, 28/30 (93.3%) in patients with genotype 1a infection, and 32/34 (94.1%) in cirrhotic patients (Table 2B/Figure S1).
Pretreatment RAVs and virologic outcome
In this cohort of patients previously exposed to licensed protease inhibitors, baseline NS3 variants commonly associated with resistance to earlier-generation protease inhibitors were found in 34 (43.6%) of the 78 evaluable patients by population sequencing (Table S1). At entry, 32 (49.2%) of the 65 evaluable patients with virologic failure harbored NS3 RAVs compared to 2 (15.4%) of the 13 evaluable patients with other reasons for not achieving SVR on their earlier regimen. Patients infected with genotype 1a had a higher prevalence of baseline NS3 RAVs than patients infected with genotype 1b [23/30 (76.7%) vs. 11/48 (22.9%)]. The prevalence of RAVs was similar in patients with or without cirrhosis. The most common NS3 polymorphisms identified in patients at baseline were 36M/L, T54S, Q80K, S122G, and R155D/K/T. The frequency of individual polymorphisms varied with the infecting sub-genotype. Q80K was detected in 11/30 (36.7%) patients with genotype 1a infections.
Baseline signature NS3 RAVs conferring only ≤5-fold decreased susceptibility to grazoprevir in vitro were identified in 30 (38.4%) of the 78 evaluable patients. In addition, NS3 RAVs with >5-fold decreased susceptibility to grazoprevir were detected in 4 (5.1%) other patients. Q80 substitutions did not decrease the in vitro activity of grazoprevir. All 44 (100.0%) patients without baseline NS3 variants and 31/34 (91.2%) patients with baseline NS3 variants associated with earlier-generation protease-inhibitors achieved SVR12. Among the 34 patients with NS3 RAVs, SVR12 rates were 28/30 (93.3%) and 3/4 (75.0%), respectively, when variant replicons had grazoprevir EC50 ≤5X versus >5X relative to the EC50 for the wild-type referent stain (Table 3). Among the 11 patients with genotype 1a variants containing the Q80K polymorphism, 10 (90.9%) achieved SVR12 compared to 18 (94.7%) of the 19 genotype 1a patients without this substitution (Table S2).
Eight (10.1%) of 79 evaluable patients harbored NS5A polymorphisms at baseline; in 5 (62.5%) of these 8 cases, the NS5a variant was associated with >5x decreased in-vitro susceptibility to elbasvir. SVR12 was achieved in 6 (75%) patients with baseline NS5A variants, including 4 (66.7%) of the 6 patients with both NS3 and NS5A variants at baseline. Thus, 2 (66.7%) of the 3 failures had baseline polymorphisms detected by population sequencing in both genes.
New variants at NS3 or NS5A loci emerged after study therapy in the 3 virologic failures (Table 4). A156T (which conferred >5x increase in grazoprevir EC50 in vitro) emerged in the NS3 gene of virus from all 3 patients, while Y93H (which conferred >5x increase in elbasvir EC50 in vitro) emerged in the viral NS5A gene in 2 cases.
Safety
Therapy was generally well tolerated in this treatment-experienced population (Table 5A). The only subject that did not complete study therapy had also discontinued prior therapy due to drug intolerance. Over the course of the entire study, 5 serious adverse events (bacterial pharyngitis, laryngeal squamous cell carcinoma, chronic obstructive pulmonary disease, urinary tract infection, and appendicitis) were reported in 5 patients, all of which were considered unrelated to study drugs. In 1 of these 5 cases, the adverse experience (appendicitis) developed >14 days after study medication had been completed.
The most commonly reported specific adverse events included fatigue, headache, asthenia, and a variety of gastrointestinal complaints (Table 5B). All adverse events during and up to 14 days after study therapy were comprehensively recorded (Table S3). Cytopenia of any blood line was infrequent, and only 8 patients had documented hemoglobin levels below 10 g/dL (Table 5C). The ribavirin dose was reduced in 11 (13.9%) patients, all of whom achieved SVR12. No patients developed grade 2 through 4 elevations of serum hepatocellular enzyme levels (Table S4).
|
| |
|
|
|
|
|