 |
|
|
| |
Good Response to TAF Plus EVG/COBI/FTC in Adolescents Through 24 Weeks
|
| |
| |
Download the PDF here
Download the PDF here
16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy, May 26-28, 2015, Washington, DC
Mark Mascolini
Pharmacokinetics (PKs) of tenofovir alafenamide (TAF), elvitegravir, cobicistat, and emtricitabine (E/C/F/TAF) in adolescents who started the single-pill combination were consistent with PKs in adults, and virologic response rates through 24 weeks exceeded 90% [1].
TAF coformulated with E/C/F proved virologically noninferior to TDF coformulated with those drugs as Stribild in two 48-week phase 3 trials in adults [2]. Markers of renal and bone toxicity favored TAF over TDF in those adult trials.
An open-label single-arm phase 3 trial enrolled 48 antiretroviral-naive adolescents from 12 to under 18 years old and weighing 35 kg or more. All adolescents had a viral load above 1000 copies, a CD4 count above 100, and an estimated glomerular filtration rate (eGFR) above 90 mL/min. Researchers compared steady-state PKs in these youngsters with an adult reference population by analysis of variance. This planned interim analysis focused on results at week 24.
The 48 study participants in Uganda (63%), the United States (19%), Thailand (13%), and South Africa (6%) had a median age of 15 (range 12 to 17) and a median weight of 52 kg (range 35 to 89). Twenty-eight (58%) were girls, 88% black, 12% Asian, and 67% vertically infected. Median pretreatment viral load stood at 4.62 log10 (about 42,000 copies), and 21% of the group had a viral load above 100,000 copies. Median CD4 count measured 452, medium serum creatinine 0.57 mg/dL, and median eGFR 158 mL/min (range 101 to 284).
PK profiles of TAF, tenofovir, elvitegravir, cobicistat, and emtricitabine were consistent with those in adults. Concentrations of the component drugs reached therapeutic levels within the ranges linked to safety and antiviral efficacy in adults.
Among 23 study participants with virologic data through 24 weeks, 21 (91%) had a viral load below 50 copies and 22 (96%) had a viral load below 400 copies. Two youngsters (9%) met criteria for virologic failure (viral load above 50 copies at 24 weeks), but those 2 had a viral load below 50 copies on retesting.
The one serious adverse event that developed--visual impairment and intermediate uveitis--resolved without treatment interruption. No one stopped taking study drugs because of adverse events. Median serum creatinine rose 0.08 mg/dL through 24 weeks, reflecting inhibition of renal tubular creatinine secretion by cobicistat. No participants had renal failure or proximal renal tubulopathy.
Median spine bone mineral density (BMD) rose 2.8% through week 24, median height-adjusted z-score for spine BMD fell 0.02, and only 2 of 23 participants (9%) had a 4% or greater decrease in z-score. Median total body-less-head BMD rose 0.34% through week 24, height-adjusted z-score for total body-less-head fell 0.09, and no one had a decrease in that z-score of 4% or more. No adolescents suffered fractures.
CROI/2015: Safety, Efficacy and Pharmacokinetics of the Integrase Inhibitor-Based E/C/F/TAF Single-Tablet Regimen in Treatment-Naïve HIV-Infected Adolescents Through 24 Weeks of Treatment - (03/16/15)
References
1. Kizito H, Gaur A, Prasitsuebsai W, et al. Week 24 data from a phase 3 clinical trial of E/C/F/TAF in HIV-infected adolescents. 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy. May 26-28, 2015. Washington, DC. Abstract 36.
2. Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. Published online April 16, 2015 http://dx.doi.org/10.1016/S0140-6736(15)60616-X.
CROI: Renal and Bone Safety of Tenofovir Alafenamide vs Tenofovir Disoproxil Fumarate - (02/27/15)
CROI: Tenofovir Alafenamide (TAF) in a Single-Tablet Regimen in Initial HIV-1 Therapy - (02/26/15)
CROI: Gilead Announces Phase 3 Results for Investigational Once-Daily Single Tablet HIV Regimen Containing Tenofovir Alafenamide (TAF) - (02/27/15)
CROI: Safety of Tenofovir Alafenamide in Renal Impairment - (02/27/15)
---------------------------
The Lancet April 15 2015
"novel tenofovir prodrug, tenofovir alafenamide achieved a high rate of virological suppression when given as part of a coformulated tablet that included emtricitabine, elvitegravir, and cobicistat. The response was non-inferior to the control group, which consisted of the approved single tablet regimen of elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate. The results were mostly non-inferior between the two groups.....
Viral Results - "We noted significant differences in efficacy for those with fewer than 100 000 copies per mL baseline HIV-1 RNA (94% for E/C/F/tenofovir alafenamide vs 91% for tenofovir disoproxil fumarate, difference in percentage 3·1%, 95% CI 0·2-6·0) and for women (95% for tenofovir alafenamide and 87% for tenofovir disoproxil fumarate, difference in percentage 7·9%, 95% CI 0·2-15·6).......E/C/F/tenofovir alafenamide was non-inferior to E/C/F/tenofovir disoproxil fumarate, with 800 (92%) of 866 patients in the tenofovir alafenamide group and 784 (90%) of 867 patients in the tenofovir disoproxil fumarate group having plasma HIV-1 RNA less than 50 copies per mL (adjusted difference 2·0%, 95% CI -0·7 to 4·7)......Virological failure was infrequent in both groups, arising in 3·6% of paitents given E/C/F/tenofovir alafenamide and 4·0% of patients given E/C/F/tenofovir disoproxil fumarate."
PK- "Plasma tenofovir exposure (AUCtau) after administration of E/C/F/tenofovir alafenamide was 91% lower than tenofovir exposure achieved with administration of E/C/F/tenofovir disoproxil fumarate (appendix). The PBMC tenofovir-diphosphate AUCtau was 4·1 times higher in participants receiving E/C/F/tenofovir alafenamide than in those receiving E/C/F/tenofovir disoproxil fumarate."
Resistance - "We noted virological failure with resistance in seven (0·8%) of 866 patients in the E/C/F/tenofovir alafenamide group versus five (0·6%) of 867 patients in the E/C/F/tenofovir disoproxil fumarate group (appendix). Resistance mutation development was similar between treatment groups (appendix). All patients with emergent resistance developed the reverse transcriptase mutation, Met184Val/Ile. One patient in the E/C/F/tenofovir alafenamide group and in two patients in E/C/F/tenofovir disoproxil fumarate developed the Lys65Arg reverse transcriptase mutation. Eight of 12 patients (five in the E/C/F/tenofovir alafenamide group and three in the E/C/F/tenofovir disoproxil fumarate group) developed primary INSTI-R, all of which were genotypically susceptible to dolutegravir. We did not record any novel tenofovir resistance mutations in any of the patients given E/C/F/tenofovir alafenamide."
Adverse Events - "Both treatments were well tolerated, with most adverse events reported as mild or moderate in severity (appendix). Adverse events leading to study drug discontinuation were uncommon: E/C/F/tenofovir alafenamide 8 (0·9%) and E/C/F/tenofovir disoproxil fumarate 13 (1·5%); adverse events leading to study drug discontinuation deemed related to study drugs were similar: E/C/F/tenofovir alafenamide 7 (0·8%) and E/C/F/tenofovir disoproxil fumarate 11 (1·3%).....
......There were no discontinuations due to renal adverse events in the E/C/F/tenofovir alafenamide group. Four patients in the E/C/F/tenofovir disoproxil fumarate group discontinued study drug because of renal adverse events. Three patients had decreased glomerular filtration rate and another patient developed nephropathy, all believed to be related to study drug. We noted no cases of proximal renal tubulopathy (including Fanconi syndrome) in either treatment group. We recorded decreases from baseline in mean estimated glomerular filtration rate by week 2 with no further change thereafter. We noted significantly smaller decreases in estimated glomerular filtration rate in the E/C/F/tenofovir alafenamide group than in the E/C/F/tenofovir disoproxil fumarate group (appendix). At 48 weeks, quantitative proteinuria (total urinary protein, albumin, retinol binding protein and ß2-microglobulin to urine creatinine ratios) increased from baseline in the E/C/F/tenofovir disoproxil fumarate group; reductions or significantly smaller increases in these urinary proteins were noted in the E/C/F/tenofovir alafenamide group (figure 3). Other measures of proximal renal tubular function (fractional excretion of phosphate and uric acid) showed significantly less change in patients receiving E/C/F/tenofovir alafenamide compared with the E/C/F/tenofovir disoproxil fumarate group (data not shown)......
.......Fractures were uncommon in both treatment groups (one in the E/C/F/tenofovir alafenamide group and seven in the E/C/F/tenofovir disoproxil fumarate group), and deemed by the investigator to be the result of trauma and unrelated to the study drugs; none resulted in permanent discontinuation of study drugs. Patients in the E/C/F/tenofovir alafenamide group had significantly less reduction in bone mineral density than those in the E/C/F/tenofovir disoproxil fumarate group through 48 weeks (figure 4). Decrease in bone mineral density was significantly lower in the E/C/F/tenofovir alafenamide group for both lumbar spine (mean -1·30% [SD 3·08] vs -2·86 [3·25]; p<0·0001) and total hip (-0·66 [3·26] vs -2·95 [3·41], p<0·0001; figure 3). Roughly one-third as many patients in the E/C/F/tenofovir alafenamide had more than 3% bone loss at the hip (E/C/F/tenofovir alafenamide 131/780 [16·8%]; E/C/F/tenofovir disoproxil fumarate 384/767 [50·1%]), and about half as many patients in the E/C/F/tenofovir alafenamide group had more than 3% bone loss at the spine (E/C/F/tenofovir alafenamide 208/784 [26·5%]; E/C/F/tenofovir disoproxil fumarate 354/773 [45·8%]; appendix).......
......We recorded greater increases in the fasting lipid parameters total cholesterol, direct low-density lipoprotein, high-density lipoprotein, and triglycerides, but identical changes in total cholesterol to high-density lipoprotein ratio, in patients given E/C/F/tenofovir alafenamide compared with those given E/C/F/tenofovir disoproxil fumarate at week 48 (appendix). 31 (3·6%) of 866 of patients given E/C/F/tenofovir alafenamide and 25 (2·9%) of 867 of participants given E/C/F/tenofovir disoproxil fumarate started lipid-lowering drugs (p=0·42)."
------------------
Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials
Paul E Sax, David Wohl, Michael T Yin, Frank Post, Edwin DeJesus, Michael Saag, Anton Pozniak, Melanie Thompson, Daniel Podzamczer, Jean Michel Molina, Shinichi Oka, Ellen Koenig, Benoit Trottier, Jaime Andrade-Villanueva, Gordon Crofoot, Joseph M Custodio, Andrew Plummer, Lijie Zhong, Huyen Cao, Hal Martin, Christian Callebaut, Andrew K Cheng, Marshall W Fordyce, Scott McCallister, for the GS-US-292-0104/0111 Study Team*
Summary
Background
Tenofovir disoproxil fumarate can cause renal and bone toxic effects related to high plasma tenofovir concentrations. Tenofovir alafenamide is a novel tenofovir prodrug with a 90% reduction in plasma tenofovir concentrations. Tenofovir alafenamide-containing regimens can have improved renal and bone safety compared with tenofovir disoproxil fumarate-containing regimens.
Methods
In these two controlled, double-blind phase 3 studies, we recruited treatment-naive HIV-infected patients with an estimated creatinine clearance of 50 mL per min or higher from 178 outpatient centres in 16 countries. Patients were randomly assigned (1:1) to receive once-daily oral tablets containing 150 mg elvitegravir, 150 mg cobicistat, 200 mg emtricitabine, and 10 mg tenofovir alafenamide (E/C/F/tenofovir alafenamide) or 300 mg tenofovir disoproxil fumarate (E/C/F/tenofovir disoproxil fumarate) with matching placebo. Randomisation was done by a computer-generated allocation sequence (block size 4) and was stratified by HIV-1 RNA, CD4 count, and region (USA or ex-USA). Investigators, patients, study staff, and those assessing outcomes were masked to treatment group. All participants who received one dose of study drug were included in the primary intention-to-treat efficacy and safety analyses. The main outcomes were the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL at week 48 as defined by the the US Food and Drug Adminstration (FDA) snapshot algorithm (pre-specified non-inferiority margin of 12%) and pre-specified renal and bone endpoints at 48 weeks. These studies are registered with ClinicalTrials.gov, numbers NCT01780506 and NCT01797445.
Findings
We recruited patients from Jan 22, 2013, to Nov 4, 2013 (2175 screened and 1744 randomly assigned), and gave treatment to 1733 patients (866 given E/C/F/tenofovir alafenamide and 867 given E/C/F/tenofovir disoproxil fumarate). E/C/F/tenofovir alafenamide was non-inferior to E/C/F/tenofovir disoproxil fumarate, with 800 (92%) of 866 patients in the tenofovir alafenamide group and 784 (90%) of 867 patients in the tenofovir disoproxil fumarate group having plasma HIV-1 RNA less than 50 copies per mL (adjusted difference 2·0%, 95% CI -0·7 to 4·7). Patients given E/C/F/tenofovir alafenamide had significantly smaller mean serum creatinine increases than those given E/C/F/tenofovir disoproxil fumarate (0·08 vs 0·12 mg/dL; p<0·0001), significantly less proteinuria (median % change -3 vs 20; p<0·0001), and a significantly smaller decrease in bone mineral density at spine (mean % change -1·30 vs -2·86; p<0·0001) and hip (-0·66 vs -2·95; p<0·0001) at 48 weeks.
Interpretation
Through 48 weeks, more than 90% of patients given E/C/F/tenofovir alafenamide or E/C/F/tenofovir disoproxil fumarate had virological success. Renal and bone effects were significantly reduced in patients given E/C/F/tenofovir alafenamide. Although these studies do not have the power to assess clinical safety events such as renal failure and fractures, our data suggest that E/C/F/tenofovir alafenamide will have a favourable long-term renal and bone safety profile.
Funding
Gilead Sciences.
Introduction
Guidelines for initial treatment of HIV-1 infection recommend the use of two nucleoside reverse transcriptase inhibitors plus a third active drug from a different class.1 Of nucleoside reverse transcriptase inhibitors, tenofovir disoproxil fumarate is included in most recommended regimens. Although potent and generally well tolerated, tenofovir disoproxil fumarate can cause clinically significant renal toxic effects,2 especially in patients with risk factors for kidney disease or who are receiving concomitant ritonavir-boosted protease inhibitors.3, 4 Additionally, tenofovir disoproxil fumarate has been associated with greater reductions in bone mineral density than other antiretroviral drugs.5 In one observational study,6 investigators noted that tenofovir disoproxil fumarate exposure was associated with an increased rate of fractures.
Research in context
Evidence before this study
Although potent and generally well tolerated, tenofovir disoproxil fumarate might lead to clinically significant renal and bone disease. The risk of these side-effects is related to plasma concentrations of tenofovir. The novel tenofovir prodrug tenofovir alafenamide delivers 90% lower plasma tenofovir compared with standard tenofovir disoproxil fumarate. This pharmacology might reduce the off-target effects of tenofovir, in particular renal and bone toxicity. A phase 2 comparative trial of tenofovir alafenamide versus tenofovir disoproxil fumarate (both coformulated with elvitegravir, cobicistat, and emtricitabine [E/C/F]) showed similar efficacy of tenofovir alafenamide and tenofovir disoproxil fumarate with a significantly reduced effect on estimated glomerular filtration rate, tubular proteinuria, and bone mineral density. We did a systematic search of PubMed to explore the use of tenofovir alafenamide in treatment-naive patients, with a particular focus on renal and bone safety in treatment-naive patients. Search terms included "tenofovir alafenamide" AND "naive" AND "renal" OR "bone." Searches were limited to articles published in English between 1997 and March, 2015. Only one article was retrieved, which was the phase 2 randomised clinical trial comparing E/C/F/tenofovir alafenamide with E/C/F/tenofovir disoproxil fumarate.
Added value of this study
These two fully-powered phase 3 double-blind, international clinical trials compared single-tablet regimens of E/C/F/tenofovir alafenamide with E/C/F/tenofovir disoproxil fumarate, with results confirming the earlier findings. Both regimens showed higher than 90% efficacy, with low (<1%) rates of discontinuations due to adverse events. Compared with tenofovir disoproxil fumarate, tenofovir alafenamide treatment led to smaller decreases in estimated glomerular filtration rate, less proteinuria (significant for all types measured), and had a more favourable effect on hip and spine bone mineral density. All lipid fractions increased more in the tenofovir alafenamide than in the tenofovir disoproxil fumarate group with similar total to HDL cholesterol ratios.
Implications of all the available evidence
E/C/F/tenofovir alafenamide is a highly effective regimen for treatment-naive patients, with more favourable effects than E/C/F/tenofovir disoproxil fumarate on renal and bone health. The hope is that these findings will translate into improved safety of tenofovir alafenamide-based antiretroviral therapy over years of treatment while maintaining a similarly high efficacy rate.
As a prodrug, tenofovir disoproxil fumarate is initially metabolised to tenofovir, which is subsequently metabolised in cells to tenofovir-diphosphate. Although intracellular tenofovir-diphosphate is responsible for the drug's antiviral activity, higher circulating plasma levels of tenofovir have been associated with an increased risk of both renal and bone toxicity.7, 8, 9, 10 A novel tenofovir prodrug tenofovir alafenamide results in roughly four times higher intracellular concentrations of the active metabolite tenofovir-diphosphate compared with tenofovir disoproxil fumarate, allowing for much lower doses of tenofovir alafenamide versus tenofovir disoproxil fumarate.11 Because of tenofovir alafenamide's reduced dose and the improved stability, plasma exposure of tenofovir is 90% lower with tenofovir alafenamide than with tenofovir disoproxil fumarate, which is believed to reduce the risk of renal and bone toxicity.7
Findings of a phase 2 comparative trial12 of tenofovir alafenamide versus tenofovir disoproxil fumarate (both coformulated with elvitegravir, cobicistat, and emtricitabine) showed similar antiviral activity of tenofovir alafenamide and tenofovir disoproxil fumarate, with a significantly reduced effect of tenofovir alafenamide compared to tenofovir disoproxil fumarate on estimated glomerular filtration rate, tubular proteinuria, and bone mineral density. To confirm these findings, we did two phase 3, double-blind clinical trials comparing elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide (E/C/F/tenofovir alafenamide) with elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate (E/C/F/tenofovir disoproxil fumarate), with a protocol-specified focus on renal and bone safety.
Methods
Study design and patients
GS-US-292-0104 and GS-US-292-0111 are randomised, double-blind, multicentre, active-controlled phase 3 trials done at 134 sites in North America, Europe, Australia, Japan, and Thailand (GS-US-292-0104), and 128 sites in North America, Europe, and Latin America (GS-US-292-0111). Studies were undertaken in accordance with the Declaration of Helsinki and were approved by central or site-specific review boards or ethics committees. All patients gave written informed consent. Adults (aged ≥18 years) were enrolled if they had HIV-1 and no previous antiretroviral treatment, had HIV-1 RNA concentration of at least 1000 copies per mL, and an estimated glomerular filtration (creatinine clearance, Cockcroft-Gault) rate of at least 50 mL per min. Eligible patients had a screening HIV-1 genotype showing sensitivity to elvitegravir, emtricitabine, and tenofovir. No CD4 entry criteria were used. We excluded patients with positive hepatitis B surface antigen or hepatitis C antibody or a new AIDS-defining illness within 30 days of screening.
Randomisation and masking
Eligible patients were randomly assigned (1:1) to receive either coformulated 150 mg elvitegravir, 150 mg cobicistat, 200 mg emtricitabine, and 10 mg tenofovir alafenamide once a day, or coformulated 150 mg elvitegravir, 150 mg cobicistat, 200 mg emtricitabine, and 300 mg tenofovir disoproxil fumarate once a day. Both regimens were given with food. Patients also received placebo tablets matching the alternative treatment; thus, investigators, patients, and study staff giving treatment, assessing outcomes, and collecting data were masked to treatment group. A computer-generated allocation sequence (block size 4) was created by Bracket (San Francisco, CA, USA), and randomisation was stratified by HIV-1 RNA (≤100 000 copies per mL, >100 000 to ≤400 000 copies per mL, or >400 000 copies per mL), CD4 count (<50 cells per μL, 50-199 cells per μL, or ≥200 cells per μL), and region (USA or ex-USA) at screening. Study investigators determined eligibility, obtained a participant number, and received automated treatment assignment based on a randomisation sequence.
Procedures
Post-baseline study visits occurred at weeks 2, 4, 8, 12, 16, 24, 36, and 48, after which patients continued masked treatment with visits every 12 weeks until week 96. After the primary endpoint had been reached, masked treatment with study drug was extended to week 144. Laboratory tests included haematological analysis, serum chemistry tests, fasting lipid parameters, CD4 counts, measures of renal function (estimated glomerular filtration rate, urine protein to creatinine ratio, urine albumin to creatinine ratio, retinol binding protein to creatinine ratio, ß2-microglobulin to creatinine ratio, fractional excretion of uric acid, and fractional excretion of phosphate; Covance Laboratories, Indianapolis, IN, USA), and measurement of HIV RNA concentration (Roche TaqMan 2.0; Roche Diagnostics, Rotkreuz, Switzerland).
We used definitions of suboptimum virological response (<1 log10 reduction from baseline HIV-1 RNA and ≥50 copies per mL at the week 8 visit, confirmed at a subsequent visit) and virological rebound (plasma HIV-1 RNA <50 copies per mL, then having HIV-1 RNA ≥50 copies per mL, confirmed at a subsequent visit) to assess virological response. We defined virological failure as plasma HIV-1 RNA greater than or equal to 50 copies per mL and less than 1 log10 reduction from baseline at week 8, or 50 copies per mL or more HIV-1 RNA after previous suppression to less than 50 copies per mL or more than a 1 log10 increase in HIV-1 RNA from nadir. Any participant meeting these criteria had a second, confirmatory sample drawn within 3-6 weeks. Confirmatory samples with 400 copies per mL or more HIV-1 RNA were sent for HIV-1 genotype and phenotype analysis (PhenoSenseGT for Protease and Reverse Transcriptase genes, GenSeq Integrase and Phenosense Integrase for the Integrase gene; Monogram Biosciences, South San Francisco, CA, USA).
In all patients, dual energy x-ray absorptiometry scans of the lumbar spine and hip were done at baseline, week 24, and week 48 to measure percent changes in bone mineral density. The scans were processed by BioClinica (Newton, PA, USA). The preliminary results were reviewed twice by an independent data monitoring committee when half of patients had completed week 12 and when all patients had completed week 24 of follow-up, respectively. The primary endpoint analysis was done after all enrolled patients had completed their week 48 study visit or had prematurely discontinued study drug.
The primary endpoint was the proportion of patients who had plasma HIV-1 RNA less than 50 copies per mL at week 48 as defined by the the US Food and Drug Adminstration (FDA) snapshot algorithm.13 Four key safety endpoints were pre-specified with multiplicity adjustments: hip bone mineral density, spine bone mineral density, serum creatinine, and treatment-emergent proteinuria. Additional secondary endpoints included treatment responses by subgroups, proportion of patients with plasma HIV-1 RNA less than 50 copies per mL when classifying missing as failure and missing as excluded, patients with HIV-1 RNA less than 20 copies per mL by snapshot, and change in CD4 count from baseline.
Statistical analysis
These two phase 3 studies were combined for a pre-specified pooled efficacy and safety analysis. Within each phase 3 study, for each of two interim analyses done for the independent data monitoring committee meeting, an α of 0·00001 was spent. Therefore, the significance level for the 1-sided non-inferiority test in the primary analysis at week 48 was 0·02499, equivalent to a two-sided 95·002% CI. The percentage differences and the associated 95·002% CIs were computed with the baseline HIV-1 RNA concentration and region stratum adjusted Mantel-Haenszel proportions.14 To control for the overall type I error in the assessment of the primary efficacy endpoint and the four key safety endpoints, hypothesis testing was done in sequential order. The primary hypothesis of non-inferiority of E/C/F/tenofovir alafenamide relative to E/C/F/tenofovir disoproxil fumarate, with respect to the proportion of patients with less than 50 copies per mL of HIV-1 RNA at week 48 (as defined by the FDA snapshot algorithm) was tested first. The non-inferiority test was done at a one-sided, 0·02499 α level. If noninferiority was established, multiplicity adjustments were undertaken for the following safety endpoints with a fallback procedure15 in the sequential order given below with prespecified two-sided α levels: hip bone mineral density (α=0·02), spine bone mineral density (α=0·01), serum creatinine (α=0·01998), and treatment-emergent proteinuria (α=0·00). The adjusted α levels were dependent on the results from preceding tests. For all the four safety endpoints, two-sided superiority tests were done.
For pooled data, assessment of non-inferiority of E/C/F/tenofovir alafenamide compared with E/C/F/tenofovir disoproxil fumarate was done with a two-sided 95% CI (α level not adjusted), with a prespecified non-inferiority margin of 12%. In the snapshot analysis using full analysis set that included all participants randomly assigned and receiving at least one dose of study drug, participants with less than 50 copies per mL of HIV-1 RNA between days 294 and 377 (week 48 window) were classified as successes. Participants with missing HIV-1 RNA data for the week 48 analysis window, who discontinued study drug, or who changed treatment before week 48 were classified as failures. A sample size of 840 patients in each study provided at least 95% power to establish non-inferiority between the two treatment groups with an overall response rate of 85% for viral suppression at week 48. Sample sizes were calculated with nQuery Advisor (version 6.0).
We did a prespecified, per-protocol snapshot analysis, which included all participants who enrolled, received at least one dose of study drug, and did not meet any of the following prespecified criteria: discontinuation of study drug before week 48 or HIV RNA data missing in week 48 analysis window, and adherence in the bottom 2·5th percentile.
Change from baseline in CD4 cell count at week 48 was summarised by treatment group with descriptive statistics based on recorded, on-treatment data in the full analysis set. The differences in changes from baseline in CD4 cell count between treatment groups and the 95% CI were constructed with analysis of variance model, including baseline HIV-1 RNA and region as fixed covariates in the model.
The safety population included all randomly assigned patients who received at least one dose of study drug. All safety data are described in summary form on all data collected after the date study drug was first given and up to 30 days after the last dose of study drug, if the participant discontinued treatment. Adherence to the investigational antiretroviral regimens was computed as number of pills taken divided by number of pills prescribed. Adverse events were coded with the Medical Dictionary for Regulatory Activities (version 17.0). We used Fisher's exact test to compare treatment differences for adverse events and Wilcoxon rank sum test to compare treatment differences for continuous laboratory test results (SAS; version 9.2).
These studies were done according to protocol without significant deviations and are registered with ClinicalTrials.gov, numbers NCT01780506 and NCT01797445.
Outcomes
The main outcomes were the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL (non-inferiority margin of 12%) and pre-specified renal and bone endpoints at 48 weeks (centrally assessed). Secondary outcomes were percentage change from baseline in hip bone mineral density at week 48, percentage change from baseline in spine bone mineral density at week 48, change from baseline in serum creatinine at week 48, treatment-emergent proteinuria through week 48, proportion of participants with HIV-1 RNA lower than 20 per mL at week 48, change from baseline in CD4 cell count at week 48, percentage change from baseline in urine retinol binding protein to creatinine ratio at week 48, percentage change from baseline in urine ß2-microglobulin to creatinine ratio at week 48, percentage change from baseline in urine protein to creatinine ratio at week 48, and percentage change from baseline in urine albumin to creatinine ratio. Safety was assessed by physical examinations, laboratory tests, 12-lead electrocardiogram, and recording of adverse events. The pharmacokinetics of tenofovir alafenamide and its metabolite, tenofovir was assessed through an intensive pharmacokinetic substudy done on a non-randomised subset of patients at week 4 or 8, which included plasma sampling for tenofovir alafenamide and tenofovir and peripheral blood mononuclear cell sampling for intracellular tenofovir-diphosphate concentrations. Bioanalytical analyses of drug concentrations of tenofovir alafenamide and tenofovir in plasma and tenofovir-diphosphate in peripheral blood mononuclear cells were done by QPS (Newark, DE, USA).
Role of the funding source
The funder designed the study, collected and analysed data, interpreted the results, and helped write the report. PES and DW are investigators who had access to the analyzed data, independently interpreted the results, and helped write the report. All authors had access to the analysed data and could assess the results and conclusions. Additional information or analyses were available to any author upon request. PES, DW, SM, MWF, and AKC made the decision to submit the report.
Results
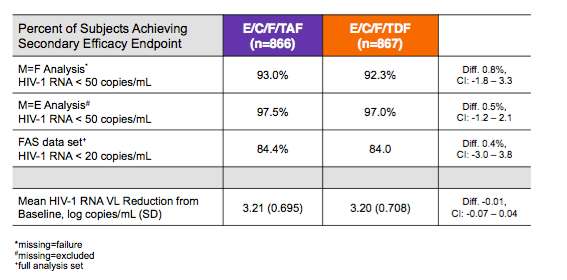
2175 patients were screened for both studies, of whom 1744 were randomly assigned to receive treatment. 1733 received at least one dose of study drug; 866 received E/C/F/tenofovir alafenamide and 867 received E/C/F/tenofovir disoproxil fumarate (figure 1). Table 1 shows baseline characteristics of participants. E/C/F/tenofovir alafenamide was non-inferior to E/C/F/tenofovir disoproxil fumarate for the combined primary outcome (800 patients [92%] vs 784 patients [90%], adjusted difference 2·0%, 95% CI -0·7% to 4·7%) and for each study (figure 2). With a cutoff of fewer than 20 copies per mL, virological outcome at week 48 by FDA snapshot algorithm was 84·4% for the E/C/F/tenofovir alafenamide group and 84·0% for the E/C/F/tenofovir disoproxil fumarate group (difference in percentages 0·4%, 95% CI -3·0% to 3·8%, p=0·83). Viral suppression was high in both treatment groups for per-protocol analysis (781 [98%] of 801 for E/C/F/tenofovir alafenamide group and 763 [97%] of 789 patients for E/C/F/tenofovir disoproxil fumarate group, adjusted difference 0·8%, 95% CI -1·0% to 2·5%) and the other the secondary efficacy endpoints (appendix) and for various subgroups (figure 2). We noted significant differences in efficacy for those with fewer than 100 000 copies per mL baseline HIV-1 RNA (94% for E/C/F/tenofovir alafenamide vs 91% for tenofovir disoproxil fumarate, difference in percentage 3·1%, 95% CI 0·2-6·0) and for women (95% for tenofovir alafenamide and 87% for tenofovir disoproxil fumarate, difference in percentage 7·9%, 95% CI 0·2-15·6). The mean increases from baseline in CD4 cell counts were higher for the E/C/F/tenofovir alafenamide group through week 48 (observed data), as follows: E/C/F/tenofovir alafenamide 230 (SD 177·3) cells per mL; E/C/F/tenofovir disoproxil fumarate 211 (170·7) cells per mL; difference in LSM 19 cells per mL, 95% CI: 3 to 36 cells per mL; p=0·024.
We noted virological failure with resistance in seven (0·8%) of 866 patients in the E/C/F/tenofovir alafenamide group versus five (0·6%) of 867 patients in the E/C/F/tenofovir disoproxil fumarate group (appendix). Resistance mutation development was similar between treatment groups (appendix). All patients with emergent resistance developed the reverse transcriptase mutation, Met184Val/Ile. One patient in the E/C/F/tenofovir alafenamide group and in two patients in E/C/F/tenofovir disoproxil fumarate developed the Lys65Arg reverse transcriptase mutation. Eight of 12 patients (five in the E/C/F/tenofovir alafenamide group and three in the E/C/F/tenofovir disoproxil fumarate group) developed primary INSTI-R, all of which were genotypically susceptible to dolutegravir. We did not record any novel tenofovir resistance mutations in any of the patients given E/C/F/tenofovir alafenamide.
36 participants in the tenofovir alafenamide group and 29 in the E/C/F/tenofovir disoproxil fumarate group participated in the intensive pharmacokinetic substudy; five of those enrolled were women. Of those, 21 patients who received E/C/F/tenofovir alafenamide and 14 who received E/C/F/tenofovir disoproxil fumarate participated in the PBMC substudy. Plasma tenofovir exposure (AUCtau) after administration of E/C/F/tenofovir alafenamide was 91% lower than tenofovir exposure achieved with administration of E/C/F/tenofovir disoproxil fumarate (appendix). The PBMC tenofovir-diphosphate AUCtau was 4·1 times higher in participants receiving E/C/F/tenofovir alafenamide than in those receiving E/C/F/tenofovir disoproxil fumarate.
Both treatments were well tolerated, with most adverse events reported as mild or moderate in severity (appendix). Adverse events leading to study drug discontinuation were uncommon: E/C/F/tenofovir alafenamide 8 (0·9%) and E/C/F/tenofovir disoproxil fumarate 13 (1·5%); adverse events leading to study drug discontinuation deemed related to study drugs were similar: E/C/F/tenofovir alafenamide 7 (0·8%) and E/C/F/tenofovir disoproxil fumarate 11 (1·3%). Table 2 shows adverse events reported by 5% or more of patients in either treatment group. Roughly 20% of patients in either group had a grade 3 or 4 laboratory abnormality (appendix). Five patients died (E/C/F/tenofovir alafenamide two patients, embolic stroke, and alcohol poisoning; E/C/F/tenofovir disoproxil fumarate three patients, cardiac arrest, multiple drug overdose, and myocardial infarction). None of the serious adverse events that resulted in the deaths were deemed related to study drugs by the investigator.
There were no discontinuations due to renal adverse events in the E/C/F/tenofovir alafenamide group. Four patients in the E/C/F/tenofovir disoproxil fumarate group discontinued study drug because of renal adverse events. Three patients had decreased glomerular filtration rate and another patient developed nephropathy, all believed to be related to study drug. We noted no cases of proximal renal tubulopathy (including Fanconi syndrome) in either treatment group. We recorded decreases from baseline in mean estimated glomerular filtration rate by week 2 with no further change thereafter. We noted significantly smaller decreases in estimated glomerular filtration rate in the E/C/F/tenofovir alafenamide group than in the E/C/F/tenofovir disoproxil fumarate group (appendix). At 48 weeks, quantitative proteinuria (total urinary protein, albumin, retinol binding protein and ß2-microglobulin to urine creatinine ratios) increased from baseline in the E/C/F/tenofovir disoproxil fumarate group; reductions or significantly smaller increases in these urinary proteins were noted in the E/C/F/tenofovir alafenamide group (figure 3). Other measures of proximal renal tubular function (fractional excretion of phosphate and uric acid) showed significantly less change in patients receiving E/C/F/tenofovir alafenamide compared with the E/C/F/tenofovir disoproxil fumarate group (data not shown).
Fractures were uncommon in both treatment groups (one in the E/C/F/tenofovir alafenamide group and seven in the E/C/F/tenofovir disoproxil fumarate group), and deemed by the investigator to be the result of trauma and unrelated to the study drugs; none resulted in permanent discontinuation of study drugs. Patients in the E/C/F/tenofovir alafenamide group had significantly less reduction in bone mineral density than those in the E/C/F/tenofovir disoproxil fumarate group through 48 weeks (figure 4). Decrease in bone mineral density was significantly lower in the E/C/F/tenofovir alafenamide group for both lumbar spine (mean -1·30% [SD 3·08] vs -2·86 [3·25]; p<0·0001) and total hip (-0·66 [3·26] vs -2·95 [3·41], p<0·0001; figure 3). Roughly one-third as many patients in the E/C/F/tenofovir alafenamide had more than 3% bone loss at the hip (E/C/F/tenofovir alafenamide 131/780 [16·8%]; E/C/F/tenofovir disoproxil fumarate 384/767 [50·1%]), and about half as many patients in the E/C/F/tenofovir alafenamide group had more than 3% bone loss at the spine (E/C/F/tenofovir alafenamide 208/784 [26·5%]; E/C/F/tenofovir disoproxil fumarate 354/773 [45·8%]; appendix).
We recorded greater increases in the fasting lipid parameters total cholesterol, direct low-density lipoprotein, high-density lipoprotein, and triglycerides, but identical changes in total cholesterol to high-density lipoprotein ratio, in patients given E/C/F/tenofovir alafenamide compared with those given E/C/F/tenofovir disoproxil fumarate at week 48 (appendix). 31 (3·6%) of 866 of patients given E/C/F/tenofovir alafenamide and 25 (2·9%) of 867 of participants given E/C/F/tenofovir disoproxil fumarate started lipid-lowering drugs (p=0·42).
Discussion
In these two randomised phase 3 clinical trials, we show that the novel tenofovir prodrug, tenofovir alafenamide achieved a high rate of virological suppression when given as part of a coformulated tablet that included emtricitabine, elvitegravir, and cobicistat. The response was non-inferior to the control group, which consisted of the approved single tablet regimen of elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate. The results were mostly non-inferior between the two groups irrespective of baseline demographic or clinical characteristics, although outcome was significantly better for tenofovir alafenamide in women and in those who had baseline viral loads lower than 100 000 copies per mL. CD4 cell count increases at week 48 were significantly greater in the tenofovir alafenamide group than in the tenofovir disoproxil fumarate group. Both coformulations were well tolerated and discontinuations for drug-related adverse events were rare in both study groups.
The high rates of successful treatment (92% in the E/C/F/tenofovir alafenamide group and and 90% in the E/C/F/tenofovir disoproxil fumarate group) marks the first time that both treatment groups in a fully powered comparative clinical trial exceeded the 90% threshold for virological suppression (plasma HIV-1 RNA <50 copies per mL) using the snapshot analysis at 48 weeks. Virological failure was infrequent in both groups, arising in 3·6% of paitents given E/C/F/tenofovir alafenamide and 4·0% of patients given E/C/F/tenofovir disoproxil fumarate. Although resistance to study treatment was not recorded in the E/C/F/tenofovir alafenamide group of the phase 2 trial, in these two larger studies a small percentage of patients (<1% in both groups) did develop drug resistance to some of the treatments, most commonly the nucleoside reverse transcriptase inhibitor mutation Met184Val selected by emtricitabine.
The high virological suppression recorded in these studies reinforces the extraordinary effectiveness of contemporary HIV treatment. With prolonged virological suppression, improved clinical outcomes, and longer survival,16 patients will potentially be exposed to antiretroviral agents for decades. As a result, maximising the safety of drugs used for HIV remains a high priority, and long-term renal and bone safety are important considerations. Although generally well tolerated as initial treatment, findings of several studies have shown an association between tenofovir disoproxil fumarate and kidney disease. A meta-analysis17 of prospective studies of HIV treatments showed a significantly greater loss of kidney function in patients receiving tenofovir disoproxil fumarate-based treatments versus non-tenofovir disoproxil fumarate regimens; a higher risk of acute renal failure was also noted. In a large cohort analysis from the Veterans Health Administration, tenofovir exposure was independently associated with proteinuria, rapid estimated glomerular filtration rate decrease, and the development estimated glomerular filtration rate less than 60 mL per min.18 Commonly cited risk factors for tenofovir disoproxil fumarate-related nephrotoxicity include older age, co-administration with ritonavir-boosted protease inhibitors (which further increase tenofovir plasma levels), and other comorbidities associated with renal disease.4
There is an increased prevalence of osteopenia and osteoporosis in patients with HIV infection.19 The cause is multifactorial, with both HIV disease-specific and treatment-specific effects observed. Generally, initiation of antiretroviral therapy leads to a reduction in bone mineral density,20 possibly related to immune reconstitution.21 This effect is larger in patients receiving tenofovir disoproxil fumarate and certain protease-inhibitor based regimens.5, 22 The mechanism of tenofovir disoproxil fumarate-related reductions in bone mineral density is poorly understood but might include osteomalacia as a result of increased urinary phosphate loss.23
In these two clinical trials, protocol-specified renal and bone endpoints confirmed the favourable safety and tolerability profile of tenofovir alafenamide reported in earlier studies. Although no particpants had overt renal failure or clinically significant tubulopathy, patients given tenofovir alafenamide had smaller reductions in estimated glomerular filtration rate and more favourable changes in urine protein to creatinine and urine albumin to creatinine ratios. Specific markers of proximal renal tubular dysfunction, including urinary retinol binding protein, urinary ß2-microglobulin, fractional excretion of uric acid, and fractional excretion of phosphate all significantly favoured the tenofovir alafenamide over the tenofovir disoproxil fumarate group, suggesting a lower potential for nephrotoxicity with tenofovir alafenamide than with tenofovir disoproxil fumarate.
The present studies represent the largest bone mineral density dataset in patients with HIV up to now. Treatment with E/C/F/tenofovir alafenamide resulted in significantly smaller reductions in bone mineral density at both the hip and the lumbar spine at week 48. The magnitude of bone mineral density decline recorded in the tenofovir alafenamide group at the hip (0·7%) similar to that seen in randomised studies of treatment-naive patients on nucleoside or nucleotide-sparing regimens.24, 25 Furthermore, with a 3% threshold for the least significant change to account for the imprecision of repeat dual energy x-ray absorptiometry measures,26 27% of patients in the tenofovir alafenamide group versus 46% in the tenofovir disoproxil fumarate group exceeded this threshold at the spine, and 17% versus 50% at the hip.
Treatment with tenofovir disoproxil fumarate has consistently been associated with less increase in lipids compared with other regimens in treatment-naive patients. The independent effect of tenofovir on lipids was most clearly shown in a study that added tenofovir disoproxil fumarate to stable background treatment in virologically suppressed patients;27 findings showed a significant reduction in total, LDL, and non-HDL cholesterol levels. In both the phase 2 comparative study of tenofovir alafenamide vs tenofovir disoproxil fumarate and the larger phase 3 studies presented here, increases in total, LDL, and HDL cholesterol, and triglycerides, were greater in the tenofovir alafenamide than the tenofovir disoproxil fumarate group. However, the difference in total cholesterol to HDL ratio at week 48 was not significantly different between treatment groups, and a small and similar proportion of participants (<4%) initiating lipid-modifying agents.
The net favourable effects on renal and bone parameters for tenofovir alafenamide almost certainly relates to the lower plasma levels of tenofovir recorded in those receiving tenofovir alafenamide instead of tenofovir disoproxil fumarate. In a pharmacokinetic substudy, plasma tenofovir exposure was 90% lower in the tenofovir alafenamide than in the tenofovir disoproxil fumarate group. Conversely, the intracellular concentration of the active metabolite, tenofovir diphosphate, was four times higher. The ability to achieve higher intracellular concentrations enables a markedly lower daily dose of tenofovir alafenamide (10 mg with ritonavir or cobicistat) versus tenofovir disoproxil fumarate (300 mg) while achieving a similar or greater antiviral effect.11 This lower dose of tenofovir alafenamide will help with both a broader range of coformulations and reduce the cost of manufacturing of the compound, the latter an important consideration in resource-limited settings. In addition to the coformulation E/C/F/tenofovir alafenamide, tenofovir alafenamide is being studied in various fixed dose combinations for HIV (with emtricitabine, with rilpivirine and emtricitabine, and with darunavir, cobicistat, and emtricitabine), and as a single agent for hepatitis B virus.
Strengths of these two studies include the large overall sample size, the randomised blinded study design with one variable of tenofovir alafenamide verus tenofovir disoproxil fumarate, and protocol-specified renal and bone endpoints. Additionally, study sites were geographically diverse, as was the ethnic origin of the participants enrolled. Limitations include a low power to assess rare clinical safety events such as renal adverse events and fractures in patients with limited baseline risk factors for kidney and bone disease, a small proportion of study participants with advanced HIV disease, a small proportion of women participants, and the exclusion of patients with chronic hepatitis B virus infection. The efficacy of tenofovir alafenamide in the treatment of chronic hepatitis B monoinfection, as well as HIV and hepatitis B virus co-infection, is currently being studied. Additionally, a clinical trial of women (NCT01705574) will give substantially more information about the efficacy, tolerability, and pharmacokinetic parameters of tenofovir alafenamide in women with HIV. Importantly, the efficacy of tenofovir alafenamide alone, or in combination with emtricitabine, for prevention, such as pre-exposure prophylaxis, is unknown and currently being explored.
In summary, in these two randomised clinical trials, treatment with a coformulated tablet of E/C/F/tenofovir alafenamide provided non-inferior virological suppression to an already approved and guidelines-recommended tablet of E/C/F/tenofovir disoproxil fumarate. Compared with tenofovir disoproxil fumarate, the nucleotide reverse transcriptase inhibitor tenofovir alafenamide showed significantly more favourable effects on renal and bone parameters. All these effects were probably related to the markedly lower plasma concentrations of tenofovir reported with tenofovir alafenamide compared with tenofovir disoproxil fumarate. Although the long-term clinical significance of these findings is unknown, it is reasonable to expect that these results will translate into improved safety of tenofovir alafenamide-based antiretroviral therapy over years of treatment while maintaining a similarly high efficacy rate.
|
| |
|
|
|
|
|