 |
|
|
| |
The 16th International Workshop on
Clinical Pharmacology of HIV and Hepatitis Therapy
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
Jennifer J. Kiser, Pharm.D.
Assistant Professor
School of Pharmacy
University of Colorado at Denver
The 16th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy was held in Washington, DC, from May 26-28, 2015. In this report we will highlight abstracts focused on pharmacologic issues that are of broad interest or might benefit from some expert clarification. Abstracts will be discussed in two general categories: (i) the clinical pharmacology of therapy for HIV infection, and (ii) the clinical pharmacology of therapy for hepatitis infection. This report does not cover all plenaries and data presented at the Workshop. For more information and to view presentations from the meeting please visit:
http://www.infectiousdiseasesonline.com/16th-hivhep-pk-presentations/
Reports: 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy
May 26-28, 2015 Washington, DC
The Clinical Pharmacology of Therapy for HIV Infection.
Courtney V. Fletcher, Pharm.D.
DRUG DEVELOPMENT
1. TAF plasma and intracellular pharmacokinetics (abstract #6).
Custodio and colleagues reported PK data for TAF in ARV-naïve persons participating in studies to evaluate the safety and efficacy of the fixed dose combination of EVG/COBI/FTC/TAF compared with EVG/COBI/FTC/TDF (Stribild). Plasma concentrations of TFV, and intracellular concentrations of TFV-diphosphate (TFV-DP) were determined and compared between TAF and TDF administration. The plasma concentrations of TFV were 91% lower with TAF than with TDF: the AUC was 307 ng*h/mL with TAF vs. 3480 with TDF. Conversely, intracellular TFV-DP concentrations in PBMCs were 4.4-fold higher with TAF than TDF. These data with the expected commercial regimen of EVG/COBI/FTC/TAF are important because they confirm previously published data (Ruane P, JAIDS 2013;63:449-55) from a dose ranging trial of TAF (8, 25 or 40mg) given as monotherapy to HIV-infected persons.
Here is the status on the FDA approval process for TAF: Gilead has submitted two new drug applications (NDA) for TAF, one in November 2014 and a second was submitted to the FDA in April 2015. The November 2014 NDA is for approval of the single tablet combination of emtricitabine (200mg), TAF (10mg), elvitegravir (150mg) and cobicistat (150mg). The FDA has a target action date on this NDA of November 2015. This second NDA submission is for two fixed dose combinations of just emtricitabine (200mg) and TAF, in two different TAF doses of 10mg and 25mg. The 10mg TAF dose is recommended if also combined with ritonavir or cobicistat (just as in the above single tablet regimen of EVG/COBI/FTC/TAF). See: http://www.gilead.com/news/press-releases/2015/4/gilead-submits-new-drug-application-to-us-food-and-drug-administration-for-fixeddose-combination-of-emtricitabinetenofovir-alafenamide-for-hiv-treatment
2. Doravirine (abstract #5).
Doravirine is an investigational NNRTI with activity against wild type virus and viruses with NNRTI mutations (e.g. K103N and Y181C). This abstract reported an investigation of exposure-response relationships from a dose ranging study of doravirine (25, 50, 100 or 200mg) vs EFV in HIV-infected adults. The investigators from Merck reported there was no correlation among doravirine PK parameters (AUC, Cmax, Ctrough) and the week 48 plasma viral load (VL), and no difference in response by baseline VL. These investigators stated a doravirine dose of 100mg once daily was selected for evaluation in Phase 3 clinical trials.
My commentary on this abstract goes to the authors' conclusion that there was no exposure-response relationship between doravirine PK and VL response at week 48. That conclusion is pharmacologic nonsense. There is always some relationship between concentrations and effect, but this study had almost no hope of ever finding one because only the week 48 time point was selected as the virologic response marker. The 100mg dose selected for Phase 3 trials may well end up being a safe and effective dose. This work, however, does not provide a solid, pharmacologic basis for the selection of that dose.
3. Atazanavir with lower dose (50mg) ritonavir (abstract #7).
12 HIV-infected persons stable on ATV/RTV at a dose of 300/100mg had plasma concentrations of ATV and RTV measured. They then underwent a RTV dose reduction to 50mg, and 7 days later had repeat PK studies. The comparison of PK parameters gave the following ratios (50mg RTV / 100mg RTV) for ATV: AUC, 0.91; Cmax, 0.97; and Cmin, 0.72. The ratios for RTV were: AUC, 0.36; Cmax, 0.38; and Cmin, 0.43. Three subjects had ATV Cmin values of <150ng/mL while receiving the 50mg RTV dose. All participants had undetectable HIV-RNA at the beginning of the study and none experienced virologic failure. Some caveats: the sample size is very small; 5 participants were receiving TDF, which is known to reduce ATV concentrations; and the assessment period is too short to draw any conclusions about a side effect benefit or increased risk of virologic failure from a reduced dose of RTV. What can be said: first, the reduced dose of RTV does not produce bioequivalent concentrations of ATV. On average ATV trough concentrations were reduced 30%, and 3 participants (25%) had ATV trough values below a suggested threshold for ATV of 150ng/mL. Second, lower concentrations (i.e. lack of pharmacokinetic bioequivalence) does not necessarily mean that the reduced RTV dose regimen would be therapeutically non-equivalent. I believe there are persons who would do just fine with a 50mg RTV dose, and these individuals could be identified through therapeutic drug monitoring (TDM). But, to generally recommend (or gain regulatory approval) the therapeutic equivalence of a reduced RTV dose would have to be conclusively demonstrated in a clinical trial (at a cost I can't quite imagine). If a 25% rate of ATV trough concentrations <150ng/mL was confirmed within a large clinical trial when the reduced RTV dose is used, I would be concerned this would translate into some reduction in the efficacy of ATV/RTV.
Finally, it is important to note that the results of this work in HIV-infected persons are different from a 2012 publication from Spanish investigators (see Estevez J, J Antimicrob Chemother 2012;67:2013-19) in healthy volunteers. In a study of 13 individuals not infected with HIV, the AUC of ATV with a RTV dose of 50mg once daily was bioequivalent to that of RTV 100mg once daily. ATV trough concentrations were reduced approximately 22%, from 660 ng/mL to 520 ng/mL (both geometric means), but none of the participants had an ATV trough value <150ng/mL. Collectively, these two studies illustrate the importance of confirming ARV pharmacokinetic characteristics in HIV-infected persons.
B. HIV PHARMACOTHERAPY AND PATIENT MANAGEMENT
1. Physiologic based pharmacokinetic modeling (PBPK) for dose prediction.
PBPK modeling is becoming a powerful tool to predict drug doses for drug development and to optimize patient management such as by predicting drug-drug interactions and appropriate dose adjustment approaches. Abstracts #s 14 and 4 provide examples of both. In abstract #14, PBPK modeling was used to evaluate the dolutegravir (DTG) dose for patients with integrase inhibitor resistant viruses. The DTG exposure and week 24 virologic response relationship found provided the basis to predict responses with a higher DTG dose of 100mg twice daily or DTG coadministration with enzyme inducing agents that decrease DTG exposure. The various simulations collectively supported the recommendation to use 50mg of DTG twice daily in patients with integrase inhibitor resistant viruses. Abstract #4 used PBPK modeling to investigate the drug-drug interactions between ARVs and two antineoplastic agents, erlotinib and gefitinib, used for the treatment of non-small cell lung cancer. The PBPK modeling predicted that concentrations of erlotinib and gefitinib, which are both CYP3A4 substrates, would be increased in the presence of RTV and decreased if given with EFV. Suggested dose adjustment strategies for erlotinib and gefitinib when given with RTV, EFV and ETR were provided, which will need clinical confirmation. I expect the field of PBPK modeling will continue to develop and grow as an important tool in guiding drug development and dose finding.
2. Low DRV trough concentrations when given with Stribild (abstract 50).
Investigators from Canada reported DRV trough concentrations in 8 HIV-infected persons who were concomitantly taking DRV 800 mg once daily with once daily Stribild (EVG/COBI/FTC/TDF). Repetitive plasma samples obtained for therapeutic drug monitoring (TDM) showed the overall median DRV trough concentration was 0.27 mg/L, which is approximately 80% lower than the population average trough (C24) of 1.4 mg/L found with DRV/RTV, 800/100 once daily. The finding of lower DRV troughs when given with COBI vs. RTV has been previously reported. In a bioequivalence study in healthy volunteers, the DRV trough concentration was 2.02 mg/L with RTV and was 1.48 with COBI and was not bioequivalent (see Kakuda TN, J Clin Pharmacol 2014;54:949). A 35% decrease in DRV trough concentrations, as seen in the healthy volunteer study probably has little clinical significance in the ARV-naïve person. The 80% decrease found by the Canadian investigators, however, is worrisome. I suggest that clinicians using the DRV + Stribild combination proceed a bit cautiously, and if access to TDM is available, I think obtaining DRV troughs, to learn more about the PK of this combination, would be very prudent.
C. HIV REMISSION AND CURE
1. Disulfiram reactivates latent HIV in a dose-dependent manner (abstract #11).
Disulfiram has been used since the 1950s for the treatment of alcohol dependence, and for this use has a well-established safety and efficacy profile. An in vitro screening assay identified disulfiram as a compound that could activate latent HIV. A 14-day pilot study in HIV-infected persons using a disulfiram dose of 500 mg once daily has been recently published (see Spivak A. Clin Infect Dis 2014;58:883). This abstract describes a dose ranging PK and PD study of disulfiram in 30 HIV-infected persons on suppressive ART. Disulfiram doses were 500, 1000 and 2000 mg, given once daily on 3 consecutive days. PK measures were concentrations in plasma of disulfiram and its 4 metabolites (M1 through M4). The measures of HIV reactivation were cell-associated unspliced (CA-US) HIV-RNA in CD4+ T cells and plasma HIV-RNA. Disulfiram concentrations at doses of 1000 and 2000 mg increased greater than dose proportional compared with 500mg. All participants demonstrated a response to disulfiram. PD modeling demonstrated a significant concentration-response relationship. The increase in CA-US HIV-RNA was most strongly linked with concentrations of the disulfiram M2 metabolite, which the increase in plasma HIV-RNA was linked with M2 and M4 concentrations. 10% of the population exhibited a much greater response to disulfiram than predicted by the PD modeling, suggesting the interplay of non-pharmacologic mechanisms. This study provides exciting data for the reactivation of latent HIV and in my mind clearly justifies further work with disulfiram. It is important to keep in mind, however, that reactivation is just the first part of the HIV cure "shock and kill" strategy; evidence points to the need for enhanced "kill" approaches.
The Clinical Pharmacology of Therapy for Hepatitis.
Jennifer J. Kiser, PharmD
A. Antiretroviral / DAA Interactions
1. Full dose daclatasvir should be used with ritonavir-boosted darunavir and lopinavir
Based on a drug interaction study with daclatasvir and ATV/RTV in healthy volunteers (see Bifano M Antivir Ther. 2013;18(7):931-40), an assumption was made that the interaction with DRV/RTV and LPV/RTV would be of the same magnitude. Thus, a dose reduction of daclatasvir from 60mg to 30mg was studied in HIV/HCV coinfected patients on DRV/RTV or LPV/RTV in ALLY-2 (Wyles D, EASL 2015). Unfortunately, individuals receiving this lower daclatasvir dose with DRV/r had lower SVR rates than other ARV groups. Data presented at this meeting (abstract #80) investigated the pharmacokinetics of this combination in healthy volunteers. Daclatasvir concentrations were compared when given at a reduced dose (30mg) with DRV/RTV 800/100mg QD or LPV/RTV 400/100mg BID to full-dose (60mg) concentrations. The daclatasvir AUC when administered at half-dose with DRV/RTV or LPV/RTV was 30% and 42% lower, respectively relative to daclatasvir AUC with 60mg. In a separate study in HIV/HCV coinfected individuals, daclatasvir trough concentrations were compared in those receiving full dose vs. half-dose daclatasvir with once-daily ritonavir-boosted darunavir (abstract #76). Daclatasvir Ctroughs in the 38 patients receiving full dose daclatasvir were similar to historical data (175 vs. 202 ng/mL) whereas the daclatasvir Ctroughs in those receiving half-dose daclatasvir (n=12) were half the historical value (96 vs. 202 ng/mL). Based on these PK data, coupled with the clinical findings from ALLY-2, full dose daclatasivr should be used with darunavir and lopinavir.
2. Newer integrase inhibitors are compatible with many DAA
Ledipasvir increases tenofovir exposures. In individuals taking EVG/COBI/TDF/FTC or TDF with a ritonavir-boosted HIV PI, higher tenofovir exposures may increase the risk of renal toxicity. TAF produces lower plasma concentrations of tenofovir and thus less renal toxicity so it may be an attractive alternative to TDF in coinfected individuals being treated with SOF/LDV. SOF/LDV was studied with EVG/COBI/FTC/TAF in 30 healthy volunteers (abstract #71). Tenofovir plasma exposures (given as TAF) were increased 27% with SOF/LDV relative to no SOF/LDV, but the average tenofovir AUC with SOF/LDV was only about 400 ng*hr/mL which is roughly 20% of typical tenofovir exposures with TDF. LDV, SOF, and 007 AUC were increased 79%, 47%, and 48%, respectively presumably due to COBI's inhibition of P-gp and/or BCRP. Since these drugs do not have recognized concentration-dependent toxicities, these increases in exposures are not expected to be problematic. These data are very reassuring on use of TAF with SOF/LDV, but TAF will not be universally available and thus additional data are needed on the safety of ritonavir-boosted PI with TDF and SOF/LDV.
SOF/LDV was also studied with TDF/FTC/DTG in 30 healthy volunteers (#71). LDV, SOF, 007, and DTG were unaffected. The average tenofovir AUC was 4900 ng*hr/mL with SOF/LDV, which is similar to the increase in tenofovir exposures when SOF/LDV is given with TDF/FTC/RPV and slightly less than the average of ~5500 ng*hr/mL with TDF/FTC plus ATV/r or DRV/r.
3D was studied with DTG and ABC/3TC (abstract #57). ABC and 3TC were not appreciably altered with 3D. DTG was increased by 38%. DTG has a wide therapeutic index, so this effect is unlikely to be problematic, but strangely both DTG and ABC/3TC appeared to reduce paritaprevir levels. Paritaprevir troughs were reduced 34% by DTG and 27% by ABC/3TC. The mechanism for this is uncertain, but the fixed dose ABC/3TC/DTG combination was not studied; rather 3D was evaluated with DTG separately from ABC/3TC. It is unclear if the effect on paritaprevir Ctrough reductions would be additive if the combination of ABC/3TC/DTG was used and if the reduction would have clinical implications for certain patient populations. The combination will be further evaluated in the ongoing TURQUOISE-I study. DRV/RTV will also be allowed in the next phase of TURQUOISE-I. While 3D causes a 50% reduction in DRV Ctroughs, data were presented at last year's workshop (Molto, O_02) that suggested this reduction did not compromise HIV suppression. I would advise against use of this combination until data from TURQUOISE-I are available.
In a study of 12 healthy volunteers (abstract #79), DTG AUC, Cmax, and Ctau were increased 33%, 29%, and 45% by daclatasvir. DTG clearance was reduced (~25%) by daclatasvir and half-life prolonged about 3 hours. DTG exposures are still within the range observed in clinical trials of HIV infected patients.
3. Rilpivirine pharmacokinetics unaltered by grazoprevir/elbasvir
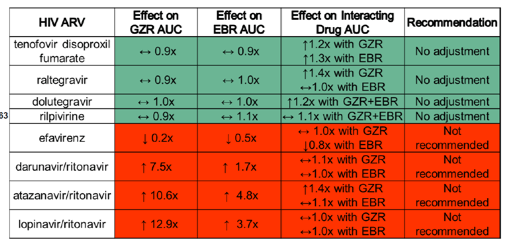
Unfortunately, ritonavir-boosted protease inhibitors and efavirenz cannot be used with grazoprevir (GZR) / elbasvir (EBR) due to significant increases and decreases in GZR/EBR concentrations, respectively, but data presented at this meeting (#63) indicate RPV can be used with GZR/EBR. A summary of ARV interactions with GZR/EBR is shown in Table 1.
Table 1. ARV interactions with grazoprevir/elbasvir (Courtesy of Wendy Yeh at Merck)
4. Newer DAA have fewer interactions with antiretroviral agents, but many patients still require a change to antiretroviral therapy or increased monitoring
There are limited data on the frequency and degree of drug interactions between DAA and ARV in practice. In readying 125 HIV/HCV coinfected patients for DAA treatment, a review of potential interactions between ARV and DAA (abstract #18) was performed at an academic medical center. Sixty four percent, 41%, 10%, and 0% of patients were on an ARV regimen that could not be used with simeprevir, 3D, sofosbuvir/ledipasvir, or daclatasvir, respectively (red in Figure 1). Seven percent, 20%, 54%, and 47% were on ARV regimens that would require increased monitoring with simeprevir, 3D, sofosbuvir/ledipasvir, and daclatasvir, respectively (yellow in Figure 1).
Figure 1. Frequency and Degree of Potential ARV Interactions with DAA in HIV/HCV Coinfected Individuals
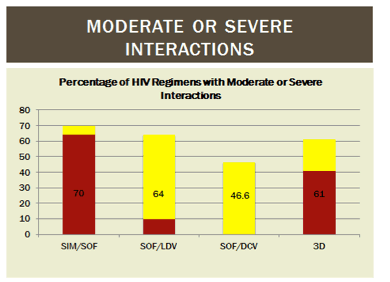
About 20% of patients either could not switch ARV therapy because they were on salvage regimens or did not want to switch ARV regimens. These data support the continued need for identification and management of potential ARV and DAA interactions in co-infected patients.
B. Interactions with DAA and other concomitant medications
1. Pravastatin can be used without adjustment with grazoprevir/elbasvir; rosuvastatin daily dose should not exceed 10mg
GZR/EBR increased pravastatin Cmax and AUC by 28% presumably due to weak inhibition of CYP3A by GZR (GZR and EBR are not OATP1B1 inhibitors). (abstract #17) GZR/EBR increased rosuvastatin Cmax and AUC by 5.49- and 2.68-fold, respectively. This interaction is the result of intestinal BCRP inhibition primarily by GZR.
2. BMS TRIO okay with a high dose oral contraceptive and SSRIs
Daclatasvir is approved in Europe and expected to receive regulatory approval as a single agent in the United States in the near future. A combination regimen (known as TRIO) that includes daclatasvir plus the HCV protease inhibitor asunaprevir and the NS5B non-nucleoside polymerase inhibitor, beclabuvir is in clinical development. TRIO was suspected to decrease ethinyl estradiol exposures based on prior data, so a "higher dose" oral contraceptive was studied (which included 1500mcg norethindrone acetate and 30mcg ethinyl estradiol). Exposures of ethinyl estradiol and norethindrone were within the therapeutic range and there was no signal of elevated liver function tests (as observed previously with 3D), thus this higher dose oral contraceptive can be used with TRIO (abstract #78).
The AUC of escitalopram and sertraline were reduced 35% and 38%, respectively by TRIO (abstract #81). This interaction is likely the result of CYP2C19 induction by asunaprevir. The SSRIs do not require a priori dose adjustments since these drugs are typically titrated to effect, but patients should be made aware of the interaction so they can discuss increased monitoring (and potential dose escalation if needed) with the SSRI prescriber.
3. 3D and 2D interactions
The potential for interactions with 3D and 8 concomitant medications with a high frequency of use in a United States commercial claims database (Lauffenburger JC, Eur J Gastroenterol Hepatol. 2014 Oct;26(10):1073-82) were evaluated (abstract #16). The results of these interaction studies are shown in Table 2.
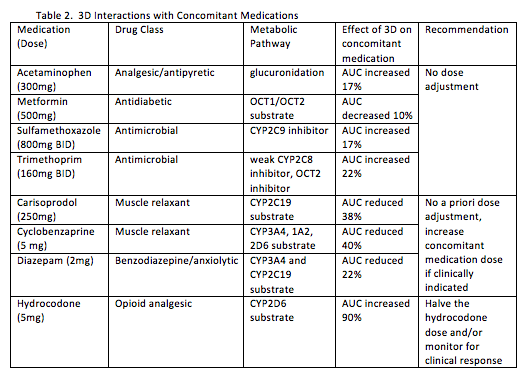
3D without dasabuvir (known as 2D) is being investigated for individuals with HCV genotype 1b and 4. Dasabuvir is an inhibitor of UGT1A1, OATP1B1 and BCRP so substrates for this enzyme or these transporters may be less affected by 2D. As with 3D, 2D does not interact with methadone or warfarin (abstract #55). Other interactions are of a similar (ketoconazole, omeprazole, pravastatin, duloxetine), smaller (rosuvastatin, buprenorphine) or greater (escitalopram, digoxin) magnitude than what is observed with 3D. As with 3D, doses of pravastatin, rosuvastatin, digoxin, and ketoconazole should be reduced when given with 2D.
C. Nucleos(t)ide Pharmacology and Pharmacokinetic-Dynamic Associations
1. Higher ribavirin exposures associated with greater likelihood of SVR in genotype 3 patients treated for 12 or 16 weeks with SOF/RBV
SVR rates with 12 or 16 weeks of SOF/RBV in genotype 3 patients are inferior to those obtained with 24 weeks of treatment, but the contribution of ribavirin exposures to the variable SVR rates has not been examined. The contribution of dose-adjusted ribavirin AUC to the likelihood of SVR in 338 genotype 3 patients receiving SOF/RBV for 12 or 16 weeks in the FISSION, POSITRON, and FUSION trials was examined (abstract #2). RBV AUC was associated with the likelihood of achieving SVR. Individuals with RBV AUC at the 75th percentile were twice as likely to achieve SVR as those individuals with RBV AUC at the 25th percentile. There is clinical utility in understanding the exposure-response relationships for ribavirin in the context of a modern HCV treatment. Weight-based ribavirin dosing was critical to optimize SVR when used with interferon, so this same dosing strategy has been applied when ribavirin is combined with DAA, but perhaps this isn't the best dosing strategy with DAA. Studies like this will inform the ribavirin dosing requirements with individual DAA combinations.
2. Intracellular sofosbuvir kinetics in red cells
The pharmacologically active form of SOF is the intracellular 007 triphosphate (007-TP or GS-461203). Measuring 007-TP in hepatocytes is logistically challenging. Peripheral blood mononuclear or red blood cells, which represent easily accessible cell alternatives, may provide insights into the clinical pharmacology of the active form of the drug. Samples from the NIAID SPARE trial (Osinusi A, JAMA. 2013;310(8):804-11) which evaluated 24 weeks of SOF with low (600mg daily) or weight-based (1000 or 1200mg daily) RBV in HCV infected individuals with genotype 1, were used to determine 007-TP concentrations in red blood cells (abstract #8). The 007-TP half-life was estimated at 69 hours. Men had ~42% lower 007-TP concentrations and a positive correlation was observed between 007-TP and ribavirin triphosphate concentrations. Average 007-TP concentration was higher, though not statistically (p=0.13), in those that achieved SVR24 vs. those that relapsed.
3. Sofosbuvir pharmacokinetics in HCV seropositive persons with end stage renal disease receiving hemodialysis
SOF is primarily renally eliminated. There are no dosing guidelines on the use of SOF in patients with CrCl <30 mL/min and those receiving hemodialysis. Thirteen HCV-infected patients requiring hemodialysis were randomized to receive SOF QD after hemodialysis or 400mg thrice weekly (abstract #19). In addition to the SOF, individuals received either daclatasvir (9 patients), simeprevir (2 patients), ledipasvir (1 patient), or RBV (1 patient). Samples were collected before hemodialysis, at the end of a 4 hour dialysis session and 1.5 hours after dose intake. 007 concentrations were higher in patients receiving SOF QD vs. thrice weekly at all three sampling times. The 007 pre-hemodialysis concentrations were higher than the post-dialysis concentrations. Pre-dialysis, 007 concentrations were 6033 ng/mL in those on QD SOF vs. 2394 ng/mL in those on thrice weekly SOF. These concentrations are 12- and 5-fold higher in those on QD and thrice weekly SOF compared with typical values in HCV infected individuals without renal impairment (Kirby B, Clin Pharmacokinet. EPub 2015 Mar 31). The treatment appeared well tolerated with no discontinuations. There was one grade 3 event (sepsis) that was considered unrelated to treatment. The study is ongoing so SVR data were still pending for half of the participants, but 7 had achieved at least an SVR4 with no relapses to date. SOF has a wide therapeutic index, so it is unclear what exposure could be associated with toxicities, but additional studies are needed to determine the safety and efficacy of SOF dosing strategies in patients with renal impairment. These data will be most relevant for individuals with both hepatic and renal impairment since treatment options are particularly limited for this population.
Abbreviations
2D = ritonavir-boosted paritaprevir and ombitasvir
3D = ritonavir-boosted paritaprevir, ombitasvir, and dasabuvir
007 = GS-331007, major drug related material in plasma following a SOF dose has no antiviral activity
007-TP = the active form of SOF measured in cells (also known as GS-461203)
%CV, percent coefficient of variation
ABC, abacavir
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
ATV, atazanavir
BID, twice daily
BCRP, breast cancer resistance protein
Cmax, maximum drug concentration
Cmin, minimum drug concentration
COBI, cobicistat
Ctrough, concentration immediately before the next dose
CYP, cytochrome P450 drug metabolizing enzymes
DCV, daclatasvir
DTG, dolutegravir
DRV, darunavir
EFV, efavirenz
EBR, elbasvir
EVG, elvitegravir
ETR, etravirine
FTC, emtricitabine
GZR, grazoprevir
HCV, Hepatitis C virus
3TC, lamivudine
LDV, ledipasvir
LPV, lopinavir
OATP1B1 = organic anion transporting polypeptide 1B1 (a hepatic uptake transporter)
PBMCs, peripheral blood mononuclear cells
P-gp, p-glycoprotein
PK, pharmacokinetic
PI, inhibitor of HIV protease
QD, once daily
RTV, ritonavir
RBV, ribavirin
RPV, rilpivirine
SOF, sofosbuvir
TAF, tenofovir alafenamide
TDF, tenofovir disoproxil fumarate
TFV-DP, tenofovir diphosphate
TFV, tenofovir
TDM, therapeutic drug monitoring
UGT1A1 = uridine glucuronosyl transferase 1A1
VL, viral load as measured by HIV-RNA
|
| |
|
|
|
|
|