 |
|
|
| |
Pitavastatin: a distinctive lipid-lowering drug
|
| |
| |
Download the PDF here
Leiv Ose

ABSTRACT
Addressing dyslipidemia is crucial to reducing the burden imposed by cardiovascular disease. However, many current statins have major limitations. Moreover, innovative treatments need to address non-LDL-C residual risk (which may be marked by high triglycerides, low HDL-C concentrations or raised ApoB:ApoA1 ratio) and increase the proportion of patients attaining treatment targets. Pitavastatin is a novel statin that induces plaque regression and is non-inferior to atorvastatin and, on some measures, superior to simvastatin and to pravastatin in the elderly. Pitavastatin addresses non-LDL-C risk factors, including producing reproducible and sustained increases in HDL-C levels. Both the pitavastatin molecule and the lactone metabolite undergo very little metabolism by CYP3A4 and, therefore, unlike some other statins, does not interact with CYP3A4 substrates. Pitavastatin is well tolerated. As such, pitavastatin shows distinctive pharmacokinetic and clinical profiles that should help a greater proportion of dyslipidemic patients attain their treatment goals.
Introduction
Cardiovascular disease (CVD) is a major contributor to global morbidity, mortality and disability. Indeed, the WHO estimates that CVD accounted for 29% of global mortality during 2004 [101]. Numerous mutually reinforcing factors, including dyslipidemia, contribute to CVD. A recent paper analyzed data from 302,430 people without vascular disease at baseline in 68 long-term prospective studies. Coronary heart disease (CHD) rates per 1000 person-years in the bottom and top thirds of baseline lipids were 2.6 and 6.2, respectively, for triglycerides, 6.4 and 2.4, respectively, for HDL-C and 2.3 and 6.7, respectively, for non-HDL-C. Adjusted hazard ratios for CHD comparing the bottom and top tertiles were 0.99, 0.78 and 1.50, respectively. CHD hazard ratios were 1.50 comparing the bottom and top tertiles for non-HDL-C:HDL-C ratio, 1.49 for ApoB:ApoA1 ratio and 1.38 for LDL-C. Hazard ratios for ischemic stroke were 1.02 comparing the bottom and top thirds for triglycerides, 0.93 for HDL-C and 1.12 for non-HDL-C [1]. Clearly, managing dyslipidemia is critical to reduce the human, clinical and societal burdens imposed by CVD.
HMG-CoA reductase inhibitors (statins) are the mainstay of dyslipidemia management. A meta-analysis of 14 studies estimated that the mean LDL-C reduction following 1 year of statin treatment varied from 0.35 to 1.77 mmol/l (mean: 1.09). All-cause and coronary mortality declined by 12 and 19%, respectively, for each mmol/l reduction in LDL-C concentrations. Furthermore, each mmol/l decline in LDL-C level was associated with a risk reduction for myocardial infarction or coronary death of 23%, coronary revascularization of 24% and fatal or nonfatal stroke of 17%. Combining these end points, each mmol/l decline in LDL-C concentration was associated with a reduction in total risk of major vascular events of 21%. The reduced risk attained statistical significance within the first year and the size of the benefit increased subsequently. Overall, 48 and 25 fewer participants with and without CHD, respectively, at baseline would experience major vascular events for every 1000 people treated with statins [1].
Despite this clinical efficacy, current statins have limitations. First, for example, there are adverse events associated with statins, including myopathy, gastrointestinal disturbances, altered liver function tests, sleep disturbances, headache, paresthesia and hypersensitivity reactions [102]. The risk of rhabdomyolysis (which, while rare, remains possibly the most serious adverse event associated with statins) is 0.44 per 10,000 treatment-years for simvastatin, atorvastatin and pravastatin. The risk reached 5.34 per 10,000 treatment-years with cerivastatin [2], which was withdrawn from the market.
Several factors contribute to the risk of developing myopathy during statin treatment. For example, the risk of developing muscular disorders with statins is sixfold higher among patients taking concurrent drugs that inhibit CYP3A4 compared with controls [3]. CYP3A4 metabolizes lovastatin, simvastatin and atorvastatin [4]. As discussed later, statins that act as a substrate for CYP3A4 potentially cause clinically significant interactions with concurrent medications and dietary components metabolized by this isoenzyme.
In fully adherent patients, statins potentially reduce LDL-C concentrations by at least 1.5 mmol/l and, therefore, the risk of major vascular events by approximately a third [5]. However, considerable residual risk (approximately two-thirds) remains in patients in whom statins reduce LDL-C levels to target values. Therefore, innovative treatments for dyslipidemia need to address a wider range of risk factors than LDL-C alone, including increased triglyceride concentrations, reduced HDL-C concentrations and increased ApoB:ApoA1 ratio. For example, among statin-treated patients with known CHD, the ApoB:ApoA1 ratio predicts clinical outcomes after correcting for standard risk factors. The corrected LDL-C:HDL-C ratio did not show this correlation [6]. An increased ApoB:ApoA1 ratio may offer a marker for early atherosclerosis as well as unstable plaques that produce weak ultrasound signals [7]. Such associations might underlie the prognostic value offered by the ApoB:ApoA1 ratio in addition to conventional risk factors.
Finally, many patients at high-risk of developing CVD, or with overt disease, have LDL-C levels that exceed those recommended in primary and secondary prevention guidelines, even when taking statin therapy. In one study, approximately half of patients did not achieve LDL-C targets with their initial statin dose. Of these, 86% had not reached the LDL-C target after 6 months, despite dose titration and receiving the clinician's statin of choice [8]. In another study, 34.7 and 27.4% of general practice patients in the UK did not attain the total cholesterol and LDL-C goals, respectively, set by Joint British guidelines within 1 year of starting statins. Furthermore, 68.2 and 57.6% of subjects failed to attain optimal levels of HDL-C and triglycerides, respectively, as defined in European management guidelines [9].
As these limitations suggest, there is still a need for new agents to manage dyslipidemia. This review examines pitavastatin, a novel statin that potentially represents an important addition to the cardiovascular armamentarium. Kowa launched pitavastatin in Japan during September 2003 for hypercholesterolemia and familial hypercholesterolemia after completing a Japanese development program. In June 2008, Kowa launched pitavastatin in Korea and Thailand [10]. Regulatory submissions have been made in a number of additional countries following the completion of a European and American development program. The US FDA approved pitavastatin doses of 1-4 mg in August 2009, and pitavastatin is currently under evaluation in Europe. The review summarizes the evidence that pitavastatin is efficacious and well tolerated in a broad range of patients and offers a distinctive pharmacodynamic and pharmacokinetic profile.
Introduction to pitavastatin & experimental data
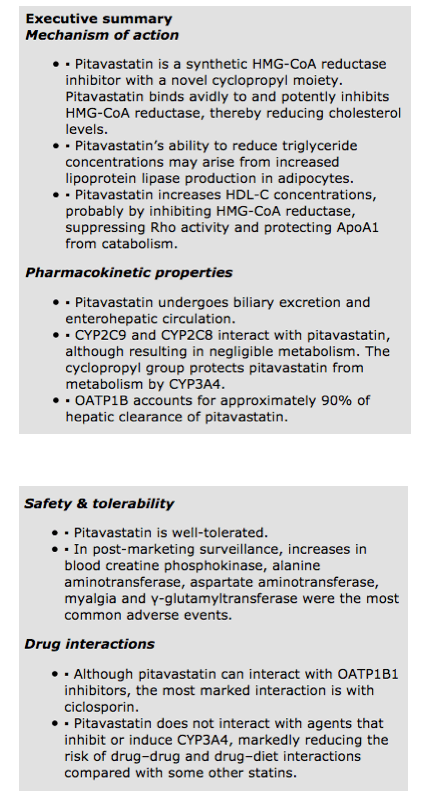
HMG-CoA reductase catalyzes mevalonate production from HMG-CoA. Pitavastatin is a synthetic HMG-CoA reductase inhibitor with a novel cyclopropyl moiety. This structural innovation means that pitavastatin binds avidly to and potently inhibits, HMG-CoA reductase. The structural similarity of statins to HMG-CoA, the precursor for cholesterol synthesis, translates into an affinity for the enzyme's catalytic pocket that is at least 1000-fold higher than the affinity of HMG-CoA reductase for the endogenous substrate [11,12].
Despite certain structural similarities, some physicochemical profiles (notably lipophilicity) vary markedly between statins (Figure 1). The ability of statins to inhibit HMG-CoA reductase is largely independent of the drug's physicochemical properties. However, membranes of the endoplasmic reticulum express HMG-CoA reductase. Physiochemical properties may influence both of the routes statins use to cross plasma membranes. First, statins diffuse passively from intracellular fluid, across plasma membranes and into the cytosol. The rate and extent of passive diffusion is, partly, a function of lipophilicity [13].
Second, an active transporter translocates statins into hepatocytes. Statins' physicochemical properties partly determine the affinity for this active transport system for the drug [13]. As discussed later, pitavastatin is a substrate of organic anion-transporting polypeptide 1B (OATP1B). Several tissues, including hepatic sinusoidal membranes, express members belonging to the OATP1B superfamily. OATP1B1 mediates translocation of pitavastatin, rosuvastatin, pravastatin, atorvastatin, fluvastatin and probably lovastatin [14-16].
Against this background, in vitro studies indicate that pitavastatin potently inhibits HMG-CoA reductase. Indeed, in cell culture, pitavastatin competitively inhibits HMG-CoA reductase 2.4- and 6.8-times more potently than simvastatin and pravastatin, respectively [17]. In a human cell line (HepG2), pitavastatin inhibited cholesterol synthesis 2.9- and 5.7-times more potently than simvastatin and atorvastatin, respectively [18]. Furthermore, Saiki and colleagues examined lipoprotein lipase expression in 3T3-L1 preadipocytes following exposure to pravastatin, simvastatin, atorvastatin and pitavastatin (1 µM for 3 days) [19]. Pitavastatin increased lipoprotein lipase activity by 30%; a greater increase than that produced by the other statins. Pitavastatin also induced strong expression of lipoprotein lipase and its mRNA. Adding mevalonate (10 µM for 3 days) weakened lipoprotein lipase activity. Thus, pitavastatin's effects on triglycerides as observed in clinical studies (vide infra) may arise from increased lipoprotein lipase production in adipocytes.
In clinical studies, pitavastatin consistently produces a marked and sustained increase in HDL-C concentrations. Indeed, pitavastatin's ability to elevate HDL-C levels is one factor differentiating it from other statins. ApoA1 is the main protein component of HDL-C. Therefore, secretion of ApoA1 is a rate-determining step in HDL production. In HepG2 cells, pitavastatin is especially potent at inducing ApoA1 (3 µM) compared with both simvastatin (10 µM) and atorvastatin (30 µM). Adding mevalonate prevented ApoA1 induction by statins. This suggests that a statin's action on ApoA1 secretion depends on inhibition of HMG-CoA reductase [20].
However, other actions may contribute to the increase in HDL-C levels produced by pitavastatin. For example, in HepG2 cells, pitavastatin increased expression of mRNA encoding the ATP-binding cassette transporter ABCA1, also known as the cholesterol efflux regulatory protein (CERP). ABCA1 controls the export of cholesterol and phospholipids, which incorporate into ApoA1 and ApoE. Again, increased ABCA1 expression depended on HMG-CoA reductase inhibition [20].
The study also examined expression of Rho and Rho kinase, which are proteins that contribute to intracellular signaling pathways. Rho and Rho kinase inhibitors (C3T and Y27632) increased ApoA1 production in HepG2 cells. Taken together, these results suggest that pitavastatin may promote ApoA1 production by three inter-related actions. First, pitavastatin inhibits HMG-CoA reductase and, second, suppresses Rho activity. Third, pitavastatin seems to protect ApoA1 from catabolism by inducing ABCA1 and augmenting lipidation (covalent binding of lipids to peptides) of ApoA1 [20].
Pharmacokinetics
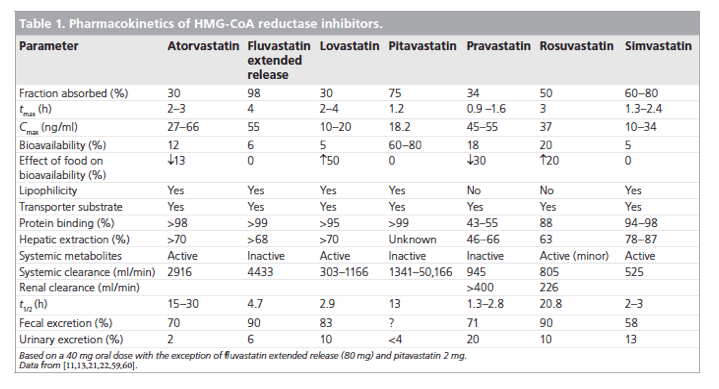
Statins show several clinically significant pharmacokinetic differences. For example, systemic bioavailability ranges from 5% with simvastatin, lovastatin and fluvastatin to more than 50% with pitavastatin (Table 1). The extent of first-pass metabolism and variations in the activity of intestinal and hepatic transport proteins apparently contribute to these differences in bioavailability. Furthermore, protein binding varies from more than 95% for pitavastatin, simvastatin, atorvastatin and lovastatin, to 50% for pravastatin [21,22].
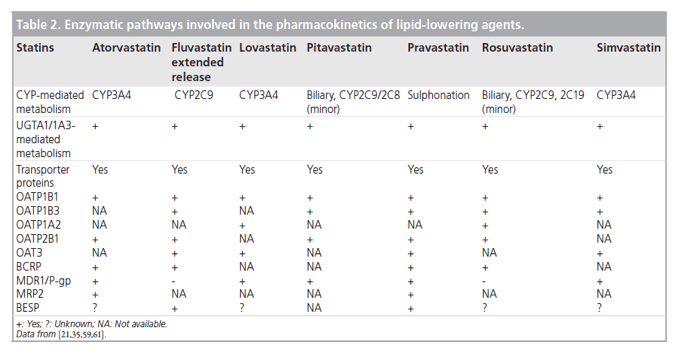
Nevertheless, the most clinically significant pharmacokinetic differences arise from variations in the metabolic and excretory pathways. Most of the bioavailable fraction of an oral dose of pitavastatin is excreted unchanged in the bile and pitavastatin undergoes enterohepatic circulation. Less than 5% of a dose of pitavastatin is excreted in the urine [22]. This pathway contrasts with those statins that undergo extensive metabolism by CYP450 isoenzymes. For example, lovastatin, simvastatin and atorvastatin are substrates for CYP3A4, and fluvastatin and rosuvastatin are metabolized by CYP2C9 [22]. The first-pass hepatic metabolism of fluvastatin varies from 50% (40 mg) to 80% (2-5 mg) [23]. Furthermore, a study using human hepatic microsomes suggested that rosuvastatin reduced the activity of CYP2C9 by 10% [24]. The cyclopropyl group on the pitavastatin molecule, which accounts for the potency of the molecule appears to 'protect' pitavastatin from metabolism by CYP3A4. Therefore, pitavastatin appears to have less potential for interactions compared with statins extensively biotransformed by CYP3A4 (vide infra) [22].
In common with most statins, pitavastatin is administered orally as an active acidic form. Glucuronosyltransferase (UGT) biotransforms open acid forms of statins. The products of UGT biotransformation are very unstable and rapidly convert to the lactone metabolite. The lactone form of many statins then undergoes rapid metabolism by CYP450 isoenzymes (Table 2). For example, the metabolic clearance catalyzed by CYP3A4 of the lactone metabolites of atorvastatin, simvastatin, cerivastatin and rosuvastatin is between 30- and 71-fold higher than biotransformation of the acid form of these statins [22]. By contrast, after administration of 2 mg/day pitavastatin in humans for 5 days, the parent compound and the lactone metabolite are the major plasma components, suggesting that the lactone form does not undergo further metabolism [25].
Transporter molecules also contribute to statins' pharmacokinetic profile. For example, p-glycoprotein shares several substrates with CYP3A4, including several statins. While, p-glycoprotein does not contribute to the bioelimination of pitavastatin, it is (as discussed previously) a substrate of OATP1B.
Hepatocytes express efflux transporters, such as breast cancer resistance protein (BCRP), which contribute to the biliary excretion of pitavastatin (Figure 2) [26]. BCRP is responsible for the efflux of several statins, including unchanged pitavastatin and rosuvastatin. A study with rosuvastatin suggests that reduced BCRP activity due to polymorphism may be associated with enhanced lipid-lowering efficacy; variability in statin efficacy with rosuvastatin, but not simvastatin, is related to BCRP polymorphism [27]. These pharmacokinetic differences influence the risk of drug-drug and drug-diet interactions.
Drug-drug & drug-diet interactions
Pitavastatin is eliminated from the liver in an unchanged form and is an important underlying reason for the low potential of drug-drug interactions. As described earlier, pitavastatin's major route of biotransformation is lactionization to an inactive metabolite. In contrast to statins such as simvastatin, lovastatin and atorvastatin, the cyclopropyl group of pitavastatin protects against metabolism by CYP3A4. Combined with the inactivity of the lactone form, pitavastatin does not show clinically significant interactions with CYP3A4 inhibitors. For example, concurrent grapefruit juice (a CYP3A4 inhibitor) increased the mean area under the concentration-time curve (AUC0-24) of atorvastatin by 83%. By contrast, concurrent grapefruit juice increased the mean AUC0-24 of pitavastatin by 13% [28]. Similarly, coadministration of itraconazole (a CYP3A4 inhibitor) produced no clinically relevant effect on the pharmacokinetics of pitavastatin or the lactone metabolite [29].
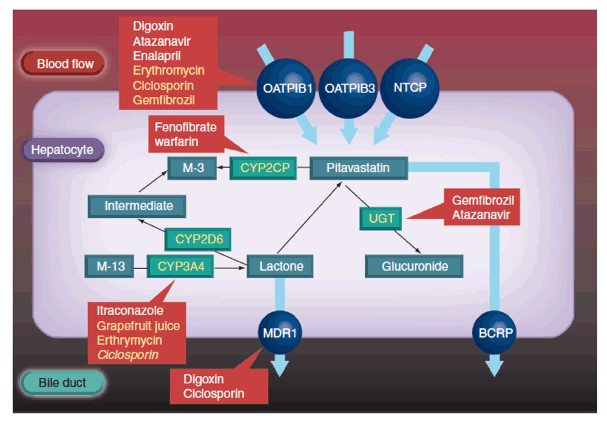
Turning to other lipid-lowering medications, pitavastatin does not show clinically significant interactions with bezafibrate or ezetimibe. Fenofibrate increased pitavastatin's steady state AUC0-24 by 18%. Gemfibrozil increased pitavastatin's steady state mean AUC0-24 and Cmax by 45 and 31%, respectively, and decreased steady state mean AUC0-24 and Cmax for the lactone metabolite by 15 and 28%, respectively. However, concurrent administration of pitavastatin and either fenofibrate or gemfibrozil was safe and well tolerated, suggesting the changes in pharmacokinetics were not clinically significant [30].
Pitavastatin undergoes only a small degree of metabolism by CYP2C9 and almost negligible transformation by CYP2C8 (Table 2) [31]. Drugs that inhibit CYP2C9 and either inhibit or compete for p-glycoprotein do not significantly interact with pitavastatin [21]. For example, digoxin is a substrate for p-glycoprotein. There is, therefore, a risk of interaction between digoxin and p-glycoprotein inhibitors, which could complicate management of atrial fibrillation, atrial flutter and heart failure. Interactions mediated by p-glycoprotein could be responsible for the increased risk of rhabdomyolysis observed among people taking combinations of some statins and digoxin. Both simvastatin and atorvastatin potentially interact with digoxin [32,33].
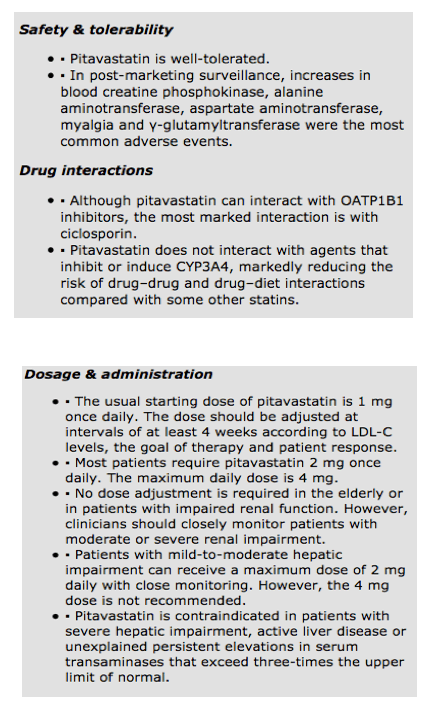
This metabolic pathway means that pitavastatin is relatively devoid of drug-drug interactions. However, there is a theoretical risk of interaction between pitavastatin and established OATP1B1 inhibitors. Gemfibrozil, which inhibits OATP1B1, increases the AUC of cerivastatin 5.6-fold, lovastatin 3.8-fold, simvastatin acid 2.9-fold, pravastatin 2.2-fold and rosuvastatin 1.9-fold [34]. However, the AUC for pitavastatin increased 1.4-fold with concurrent gemfibrozil [35], suggesting that pitavastatin is not as dependent on OATP1B1 transportation as other statins. On the other hand, coadministration of pitavastatin and ciclosporin increased mean Cmax and AUC0-24 of pitavastatin 6.6- and 4.6-fold, respectively [36]. Concurrent treatment was well tolerated by healthy adults [36].
However, ciclosporin inhibits several members of the OATP family as well as other transporters, such as p-glycoprotein [37,38].Further studies need to investigate whether these various actions account for greater interaction between ciclosporin and pitavastatin than observed between pitavastatin and other OATP inhibitors, such as fenofibrate and gemfibrozil [30]. While the pharmacokinetic interaction between ciclosporin and pitavastatin is less marked than with some other statins, there are not enough clinical data to define the safety of the coadministration of the two drugs. Therefore, concomitant use is currently contraindicated.
Overall, the most significant pharmacokinetic differences between the statins are due to differences in metabolism and excretion. Pitavastatin has a relatively low drug-drug interaction profile, although there is a speculative risk of interactions between pitavastatin and established OATP1B1 inhibitors, such as ciclosporin. Nonetheless, pitavastatin may present less potential for interactions and associated adverse events compared with those statins extensively metabolized by CYP3A4, such as lovastatin, simvastatin or atorvastatin [22,39].
Clinical data
A large and growing number of studies show that pitavastatin is clinically efficacious in the management of primary hypercholesterolemia or combined dyslipidemia in a wide range of patients. For example, in Japanese patients, pitavastatin (2 mg/day) and pravastatin (10 mg/day) reduced LDL-C levels by 38 and 18%, respectively, over 12 weeks [40]. This section focuses on clinical studies performed in Caucasian patients to support the FDA and European license applications. Doses of comparators selected in these studies were in the ranges recommended by their respective manufacturers and clinical data, although the authors accept that maximal doses are also available [41].
One of these studies demonstrated that pitavastatin was non-inferior to atorvastatin at reducing LDL-C concentrations [42]. The study enrolled 821 patients with primary hypercholesterolemia or combined dyslipidemia. After a 6-8-week dietary lead-in period, randomized patients received one of four treatment regimens for 12 weeks. Two groups received pitavastatin (2 mg/day) or atorvastatin (10 mg/day), and two groups received pitavastatin (2 mg/day) or atorvastatin (10 mg/day) for 4 weeks followed by forced titration to pitavastatin (4 mg/day) or atorvastatin (20 mg/day).
Over 12 weeks, pitavastatin produced a non-inferior reduction in LDL-C concentrations from baseline to end point (week 12 or last observation carried forward) compared with atorvastatin. The mean change was -37.9 and -37.8% for pitavastatin 2 mg/day and atorvastatin 10 mg/day, respectively, and -44.6 and -43.5% for pitavastatin 4 mg/day and atorvastatin 20 mg/day, respectively. Most patients reached National Cholesterol Education Program (NCEP) LDL-C targets: pitavastatin 4 mg, 77.9%; atorvastatin 20 mg, 70.6%; pitavastatin 2 mg, 56.8%; and atorvastatin 10 mg, 65.7%. The proportion attaining European Atherosclerosis Society (EAS) targets showed a similar pattern: pitavastatin 4 mg, 78.5%; atorvastatin 20 mg, 76.5%; pitavastatin 2 mg, 56.8%; and atorvastatin 10 mg, 59.8%. HDL-C levels increased from baseline by 4 and 5% with pitavastatin 2 and 4 mg, respectively, compared with 3 and 2.5% with atorvastatin 10 and 20 mg, respectively [42].
A second study compared pitavastatin and simvastatin in 857 patients with either primary hypercholesterolemia or combined dyslipidemia [43]. Patients received pitavastatin 2 mg/day or simvastatin 20 mg/day, with forced titration to pitavastatin 4 mg/day or simvastatin 40 mg/day in a similar manner to the study detailed above. Pitavastatin 2 mg reduced concentrations of LDL-C, non-HDL-C and total cholesterol more than simvastatin 20 mg. Furthermore, a greater proportion of patients treated with pitavastatin 2 mg achieved the EAS LDL-C target than with simvastatin 20 mg: 59.6 and 48.6% respectively, a statistically significant difference. For example, pitavastatin 2 mg and simvastatin 20 mg reduced LDL-C levels by 39.0 and 35.0%, respectively; another statistically significant difference. Pitavastatin 4 mg was non-inferior to simvastatin 40 mg. The reductions in LDL-C concentrations were 44.0 and 42.8%, respectively, while 75.2 and 75.5%, respectively, attained the EAS LDL-C target. HDL-C concentrations were increased from baseline to a similar extent by both treatments: 6.0 and 6.2% with pitavastatin 2 and 4 mg, respectively; and 5.0 and 6.8% with simvastatin 20 and 40 mg, respectively.
Of the volunteers who completed one of these studies, 1353 patients elected to receive open-label pitavastatin 4 mg once daily for up to 52 weeks. The proportion of patients achieving NCEP and EAS LDL-C targets at week 52 was 74.0 and 73.5%, respectively. Pitavastatin maintained the reduction in LDL-C that emerged during the double-blind studies. Changes in other efficacy parameters (triglycerides, total cholesterol, non-HDL-C, ApoA1 and ApoB, high-sensitivity C-reactive protein, oxidized LDL) and ratios (total cholesterol:HDL-C, non-HDL-C:HDL-C and ApoB:ApoA1) were sustained over 52 weeks compared with the end of the double-blind studies. HDL-C levels rose continually during follow-up, ultimately increasing by 14.3% over the initial baseline [44].
Overall, although these pitavastatin clinical studies demonstrate, at the least, achievement of non-inferiority criteria for LDL-C reductions versus simvastatin and atorvastatin, the clinical efficacy of pitavastatin is clearly demonstrated in attainment of EAS and NCEP targets. Patients receiving the higher dose over the 52-week, open-label interval maintained the benefits observed in the shorter-term studies. Short-term treatment with pitavastatin was not associated with increases in HDL-C levels that were significantly different from comparators. However, HDL-C concentrations increased steadily during long-term treatment with pitavastatin, at least doubling initial baseline levels after 52 weeks.
Plaque regression
Studies have yet to show that pitavastatin reduces CVD and all-cause mortality or morbidity. However, pitavastatin induces plaque regression through changes in lipid profile, pleiotropic effects or both. The JAPAN-ACS study compared 8-12 months treatment with 4 mg/day pitavastatin or 20 mg/day atorvastatin. Researchers measured coronary plaque volume in 252 patients undergoing percutaneous coronary intervention for acute coronary syndrome guided by intravascular ultrasound. LDL-C concentrations decreased by 38 and 37% from baseline, respectively. Pitavastatin and atorvastatin reduced the volumes of nonculprit plaques by 16.9 and 18.1%, respectively. In both cases, plaque regression was associated with negative vessel remodeling (113.0-105.4 mm3). The upper limit of 95% confidence interval of the mean difference in percentage change in plaque volume between pitavastatin and atorvastatin (1.11%) after adjusting for sex, diabetes and total cholesterol levels, did not exceed the 5% predefined non-inferiority margin [45].
In another recent Japanese study, the plaque volume index declined by 2.6% in patients with acute coronary syndrome treated with 80 mg/daily pitavastatin compared with a 0.2% increase in those receiving 80 mg/daily atorvastatin for 2-3 weeks [46]. The early benefit seen with pitavastatin may reflect a greater affinity for fibrofat than atorvastatin.
In part, the discordance between these studies reflects differences in the cohorts enrolled and intravascular ultrasound techniques. The Japanese study enrolled patients with acute coronary syndrome, while REVERSAL assessed the regression of stable coronary artery disease [45]. In the JAPAN-ACS study, intravascular ultrasound measurement was performed in an area adjacent to where percutaneous coronary intervention was performed. In REVERSAL, intravascular ultrasound measurement was taken from vessels where percutaneous coronary intervention had not been performed. Further studies must examine this hypothesis and determine whether plaques regress in other patient populations. However, the results are consistent with the meta-analysis demonstrating that 48 and 25 fewer participants with and without CHD, respectively, at baseline would experience major vascular events for every 1000 people treated with statins for 1 year [5]. The plaque regression studies suggest that pitavastatin may share the statins' class effect of reducing CVD-related morbidity and mortality.
Elderly patients
Elderly people are particularly likely to develop CVD. Indeed, two-thirds of first major coronary events occur in people aged 65 years or older [47]. Vascular disease is the primary cause of mortality in approximately half of this age group, while 75-80% of subjects aged 80 years show atherosclerosis [48].
Against this background, a non-inferiority study compared once-daily pitavastatin and pravastatin (1 vs 10 mg; 2 vs 20 mg; and 4 vs 40 mg) over 12 weeks in 942 patients aged 65 years or older (mean: 70; range: 65-89 years) with primary hypercholesterolemia or combined dyslipidemia and elevated plasma LDL-C levels and triglycerides. Based on mean LDL-C concentrations, pitavastatin at least met prospective non-inferiority criteria (6% non-inferiority limit) at all doses. Indeed, the mean decrease in LDL-C levels was approximately 10 percentage points greater with pitavastatin than pravastatin.
Furthermore, pitavastatin showed statistically significant benefits compared with pravastatin with: all doses for total cholesterol and ApoB; medium- and high-doses for HDL-C; and low- and high-doses for triglycerides. The proportion of patients that met the NCEP LDL-C target with pitavastatin and pravastatin, respectively, were: low doses, 83 and 65%; medium doses, 89 and 81%; and high doses, 91 and 88% [49]. This study suggests that in elderly patients, pitavastatin is more effective than pravastatin using doses that are as well tolerated.
Of the volunteers that completed this double-blind study, 545 patients aged 65 years or older entered an open-label extension phase assessing pitavastatin 2 and 4 mg once daily. During the 60-week study, adverse event rates were similar across the whole dose range of both drugs. After 60 weeks, pitavastatin reduced concentrations of LDL-C and several other atherogenic lipid parameters compared with the end of the double-blind comparative phase. HDL-C concentrations increased by 9.6% compared with the baseline for the double-blind study [49].
After 60 weeks, 99% of patients attained NCEP targets; most patients attained LDL-C targets with pitavastatin 2 mg. Patients who did not meet LDL-C targets on pitavastatin 2 mg titrated to 4 mg. Of those on the higher dose, 70% met NCEP targets at week 60. The effects of the 4 mg dose on other lipid parameters were similar to pitavastatin 2 mg [49].
Post-marketing surveillance study
Clinical studies typically include a wide range of inclusion and exclusion criteria, which potentially hinder attempts to extrapolate the results to the less-selected population encountered in clinical practice. However, a Japanese post-marketing surveillance study, known as LIVES, examined this issue. LIVES assessed pitavastatin's effectiveness and safety in 18,031 patients who were prescribed pitavastatin and registered with the study team within 2 weeks. Most patients received pitavastatin as their initial lipid-lowering therapy. However, 18.9% had previously received lipid-lowering therapy in clinical practice. Pitavastatin reduced LDL-C concentrations by 29.1% within 4 weeks of the start of treatment. LDL-C remained at this level for the remainder of the 2-year follow-up [10].
Serum LDL-C levels showed similar declines in a wide variety of patients: without and with concomitant liver disease (29.2 and 27.7%, respectively); without and with concomitant renal disease (29.1 and 28.0%, respectively); and without and with diabetes (29.7 and 27.3%, respectively). In patients with abnormal baseline triglyceride and HDL-C levels, pitavastatin decreased triglyceride concentrations by 22.7% and increased HDL-C levels by 19.9% [10]. In a further analysis of the LIVES database, concentrations of total cholesterol and LDL-C declined by 21.0 and 31.3%, respectively, over 2 years of follow-up. Triglyceride levels declined by 6.1% overall and by 24.2% in patients with baseline triglyceride levels 15.8% or higher [50].
Consistent elevation in HDL-C
As the aforementioned studies demonstrate, pitavastatin consistently produces a clinically significant increase in HDL-C levels. Other statins, in contrast, show inconsistent results on HDL-C concentrations, with elevations ranging from 0 to 12% [51]. Other studies confirm these findings. For example, an analysis of the LIVES database found that in patients with low HDL-C levels (<40 mg/dl) at baseline, HDL-C concentrations rose by 14.0 and 24.9% after 12 and 104 weeks, respectively. Indeed, HDL-C levels rose by 15.8% after patients switched to pitavastatin from other statins [50]. This suggests that patients may benefit from switching statins to pitavastatin if, ceteris paribus, HDL-C remains unacceptably low on the initial regimen.
Indeed, pitavastatin's ability to elevate HDL-C levels appears to be independent of the efficacy on other lipid outcomes. For example, a 12-week, prospective, open-label trial found comparable reductions in LDL-C concentrations with pitavastatin (2 mg/day) and atorvastatin (10 mg/day): 42.6 and 44.1%, respectively. Pitavastatin and atorvastatin also reduced levels of total cholesterol by 29.7 and 31.1%, respectively, and triglyceride concentrations by 17.3 and 10.7%, respectively. However, HDL-C levels increased significantly with pitavastatin, but not atorvastatin: 3.2 and 1.7%, respectively [52].
In a separate study, Japanese patients with LDL-C levels 140 mg/dl or greater and glucose intolerance were randomly assigned to receive either pitavastatin 2 mg/day or atorvastatin 10 mg/day in a comparative 52-week, open-label study. Increases in HDL-C levels were significantly greater following pitavastatin 2 mg/day, compared with atorvastatin 10 mg/day: 8.2 versus 2.9%, respectively. Changes in ApoAI (5.1 vs 0.6%), ApoB (-35.1 vs -28.2%) and ApoE (-28.1 vs -17.8%) also favored pitavastatin 2 mg/day versus atorvastatin 10 mg/day [53].
Safety & tolerability
Pitavastatin is well tolerated. The post-marketing LIVES study analyzed safety in 19 925 patients receiving pitavastatin in clinical practice [10]. During a 2-year follow-up, 10.4% of patients experienced adverse events, of which approximately 84% of side effects were mild and only approximately 1% were severe. Increases in blood creatine phosphokinase (2.74%), alanine aminotransferase (1.79%), myalgia (1.08%), aspartate aminotransferase and γ-glutamyltransferase (1.00%) were the most common adverse events. Only 7.4% of patients discontinued pitavastatin after developing adverse events.
Pitavastatin was also well tolerated in elderly patients. During the 2-year follow-up, there were no differences in rates of adverse events between patients under or those 65 years of age or over. In addition, regression analysis demonstrated that age (<65 vs ≥65 years) was not a significant factor for incidence of any adverse event or myopathy-associated events [10].
Furthermore, during a 52-week open-label clinical study of pitavastatin 4 mg once daily (the highest recommended dose), only 4.1% of patients withdrew due to treatment emergent adverse events. The investigators did not consider that any of the serious adverse events reported during this study were related to pitavastatin. No clinically significant abnormalities were associated with pitavastatin in routine laboratory variables, urinalysis, vital signs or 12-lead ECG. Increased creatine phosphokinase (2.74%), nasopharyngitis (5.4%) and myalgia (4.1%) were the most common treatment emergent adverse events [44].
Muscle toxicity
Muscle toxicity and in particular rhabdomyolysis, is a rare but potentially serious adverse event associated with statins. In a meta-analysis of 13 studies, the incidence of rhabdomyolysis among statin users was 0.023%. This compared with 0.015% among controls. The absolute excess risk associated with statins over 5 years (0.01%) did not reach statistical significance [5]. Similarly, the LIVES study suggested that myalgia was uncommon (1.08%) during treatment with pitavastatin. Only one patient enrolled in the trial developed rhabdomyolysis with creatine phosphokinase at least ten-times the upper limit of normal (0.005%) [10]. Furthermore, during open-label treatment with pitavastatin 4 mg once daily for up to 52 weeks, there were no reports of myopathy, myositis or rhabdomyolysis [44]. Taken together, these data suggest that pitavastatin is associated with a low risk of rhabdomyolysis. However, differences in the definition of rhabdomyolysis hinder comparisons between studies. Nevertheless, the dose of pitavastatin does not need to be adjusted in the elderly.
Dosage & administration
Patients swallow pitavastatin tablets whole at any time of the day with or without food. Ideally, patients should take pitavastatin at the same time each day. Evening doses of statins optimizes outcomes due to the circadian rhythm of lipid metabolism [54,55].
The usual starting dose of pitavastatin is 1 mg once daily. The dose should be adjusted at intervals of at least 4 weeks according to LDL-C levels, the goal of therapy and patient response. Most patients require pitavastatin 2 mg once daily. The maximum daily dose is 4 mg.
No dose adjustment is required in the elderly or in patients with impaired renal function. However, clinicians should closely monitor patients with moderate or severe renal impairment. Patients with mild-to-moderate impairment of hepatic function can receive a maximum dose of 2 mg daily with close monitoring. However, the 4 mg dose is not recommended and pitavastatin is contraindicated in patients with severe hepatic impairment, active liver disease or unexplained persistent elevations in serum transaminases that exceed three-times the upper limit of normal.
Conclusion
According to the WHO, CVD accounts for approximately a third of global mortality [101]. Managing dyslipidemia is a central to lifting this burden; each mmol/l decline in LDL-C reduces the risk of major vascular events by approximately a fifth [5]. However, current statins have limitations that hinder statin's full potential from being realized in clinical practice, including adverse events [102], residual risk from factors other than LDL-C and LDL-C levels above targets recommended in guidelines [8,9].
Pitavastatin potently inhibits HMG-CoA reductase, increases lipoprotein lipase expression in adipocytes and promotes ApoA1 production [17,19,20]. These actions lower concentrations of LDL-C and triglycerides and increase HDL-C levels, respectively. Pitavastatin undergoes biliary excretion and enterohepatic circulation; the latter may contribute to the drug's pharmacodynamic profile. Pitavastatin undergoes only a small degree of metabolism by CYP2C9 and almost negligible transformation by CYP2C8 [31]. The cyclopropyl group protects pitavastatin from metabolism by CYP3A4, reducing the risk of interactions compared with statins extensively biotransformed by this isoenzyme [22]. OATP1B is a major transporter of pitavastatin into the liver, the main site of action for statins [26]. Although pitavastatin can interact with OATP1B1 inhibitors, the most marked interaction is with ciclosporin [30,36].
In Caucasians, pitavastatin is non-inferior to atorvastatin in reducing LDL-C [42]. Over three-quarters of patients receiving pitavastatin 4 mg/day attain EAS LDL-C targets. Pitavastatin 4 mg/day is non-inferior to simvastatin 40 mg/day, respectively. Pitavastatin 2 mg reduced LDL-C, non-HDL-C and total cholesterol more than simvastatin 20 mg.
Furthermore, a greater proportion of patients taking pitavastatin 2 mg achieved the EAS LDL-C target than those receiving simvastatin 20 mg. Pitavastatin produced a sustained decline in levels of LDL-C and other lipids and maintained target attainment during long-term treatment (52 weeks). HDL-C levels rose continually during 52 weeks of treatment [44].Pitavastatin and atorvastatin produce similar reductions in plaque volume and negative vessel remodeling [45].
Patients aged 65 years or older are particularly prone to developing CVD. In these patients, pitavastatin reduced LDL-C levels by approximately 10% more than pravastatin. Indeed, pitavastatin was significantly more efficacious than pravastatin across a range of lipid outcomes. A greater proportion of the elderly patients taking pitavastatin met the EAS target compared with the pravastatin group. During a 60-week open-label extension study, pitavastatin increased HDL-C levels, reduced LDL-C concentrations and more than 90% of patients attained LDL-C targets. The post-marketing LIVES study suggests that the benefits observed in clinical studies translate into naturalistic practice.
Across the studies, pitavastatin consistently produces a clinically significant increase in HDL-C. By contrast, other statins show inconsistent results on HDL-C [51]. In theory, pitavastatin's effects on HDL-C and other lipids may offer an advantage over other statins: allowing a greater proportion of patients to attain treatment targets. However, the size and duration makes a comparative trial needed to examine this hypothesis impracticable. Pitavastatin is well tolerated. During open-label treatment with pitavastatin for up to 2 years, 7.4% of patients withdrew after developing adverse events. No clinically significant abnormalities emerged in routine laboratory variables, urinalysis, vital signs or 12-lead ECG. The most common adverse events were increased creatine phosphokinase, alanine aminotransferase, aspartate aminotransferase, myalgia and γ-glutamyltransferase [10].
Future perspective
Statins are well established in the dyslipidemia armamentarium. Future studies of pitavastatin should directly compare clinical end points with other statins to define its position within the armamentarium. The widening choice of treatments, exemplified by pitavastatin, allows clinicians to target treatment to each patient with unprecedented accuracy. In the future, the growing number of genetic polymorphisms that appear to influence outcomes in CVD should further hone accuracy.
For example, several single nucleotide polymorphisms (SNP) in genes encoding OATP1B may produce clinically relevant alterations in statin pharmacokinetics [56]. For example, nonsynonymous SNPs in SLCO1B1 (encoding influx system OATP1B1) and ABCC2 (encoding efflux system MRP2) result in several clinically significant variant alleles. One SNP (421C/A* 15/*15) produced a threefold increase in AUC0-24 of pitavastatin compared with 421C/C*1b/*1b (the wild-type in Asian populations). Another SNP (421C/C*1b/*15) doubled the AUC0-24 compared with 421C/C*1b/*1b. These functionally impaired alleles seem to be associated with a higher risk of myopathy and rhabdomyolysis [57]. However, these SNPs account for only a threefold increase in AUC0-24. Each C allele increased the odds ratio for myopathy by 4.5. Moreover, the CC genotype increased the risk 16.9-fold as compared with TT homozygotes. Indeed, the C allele accounted for more than 60% of myopathy cases. SEARCH suggests that the risk associated with SLCO1B1 polymorphisms is limited to doses of simvastatin of 40-80 mg [58].
Treatment goals may also evolve as evidence implicates a wider range of pathogenic lipids and lipoproteins. Indeed, reductions in CHD risk are proportional to the absolute decline in LDL-C. Therefore, some authors argue for achieving 'substantial absolute reductions' in LDL-C rather than aiming at a particular target [5]. Furthermore, the recognition that the factors associated with the non-LDL-C residual risk are amenable to treatment argues for therapeutic objectives that encompass a wider range of factors. While the role of statins in CHD may appear well established, there is much that clinicians and researchers can still achieve to further reduce the global burden imposed by CVD.
Financial & competing interests disclosure
Kowa Research Europe Ltd funded the study and the preparation of this manuscript. Leiv Ose received research grant funding from Merck, Pfizer, Schering-Plough, Roche and Boehringer Ingelheim; and he is a consultant/advisor to Kowa; and has received speakers bureau fees from AstraZeneca and Kowa. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
|
| |
|
|
|
|
|