| |
Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro
|
| |
| |
Download the PDF here
The JAK-STAT pathway is activated in both macrophages and lymphocytes upon human immunodeficiency virus type 1 (HIV-1) infection and thus represents an attractive cellular target to achieve HIV suppression and reduced inflammation, which may impact virus sanctuaries. Ruxolitinib and tofacitinib are JAK1/2 inhibitors that are FDA approved for rheumatoid arthritis and myelofibrosis, respectively, but their therapeutic application for treatment of HIV infection was unexplored. Both drugs demonstrated submicromolar inhibition of infection with HIV-1, HIV-2, and a simian-human immunodeficiency virus, RT-SHIV, across primary human or rhesus macaque lymphocytes and macrophages, with no apparent significant cytotoxicity at 2 to 3 logs above the median effective antiviral concentration. Combination of tofacitinib and ruxolitinib increased the efficacy by 53- to 161-fold versus that observed for monotherapy, respectively, and each drug applied alone to primary human lymphocytes displayed similar efficacy against HIV-1 containing various polymerase substitutions. Both drugs inhibited virus replication in lymphocytes stimulated with phytohemagglutinin (PHA) plus interleukin-2 (IL-2), but not PHA alone, and inhibited reactivation of latent HIV-1 at low-micromolar concentrations across the J-Lat T cell latency model and in primary human central memory lymphocytes. Thus, targeted inhibition of JAK provided a selective, potent, and novel mechanism to inhibit HIV-1 replication in lymphocytes and macrophages, replication of drug-resistant HIV-1, and reactivation of latent HIV-1 and has the potential to reset the immunologic milieu in HIV-infected individuals.
----------------------
Evaluating the Safety and Tolerability of Ruxolitinib in Antiretroviral-Treated HIV-Infected Adults
Researchers will evaluate the effect ruxolitinib has on inflammation and immune activation. [Reservoirs will be evaluated]
---------------------
Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro
"Together, these data demonstrate that targeted inhibition of the JAK-STAT pathway by inhibition of JAK provides a selective, effective, and novel mechanism to inhibit HIV-1 replication in lymphocytes and macrophages, replication of drug-resistant HIV-1, and reactivation of latent HIV-1.
Inhibition of the JAK-STAT pathway using FDA-approved agents, such as ruxolitinib and tofacitinib, provides the first opportunity to have a significant impact on this inflammation and may reset the immunologic milieu. These findings present a compelling rationale for further work designed to elucidate the potential clinical relevance of these drugs in a macaque model and eventually in humans at a safe and effective dose.
Herein, we report for the first time that two FDA-approved JAK inhibitors, ruxolitinib and tofacitinib, are effective, submicromolar inhibitors of HIV replication in lymphocytes and macrophages. .....The established anti-inflammatory profile of ruxolitinib and tofacitinib suggests that these compounds may be effective inhibitors of homeostatic proliferation and reservoir maintenance of TCM, TTM, or TSCM cells, which is driven by IL-7 and IL-15, two proinflammatory cytokines (30, 34). Concurrently, it is possible that a reduction in systemic inflammation and activation could lead to reduction of HIV-1 coreceptor expression including CCR5 as its expression is positively correlated with activation state
.....The ability of ruxolitinib and tofacitinib to inhibit IL-6 and TNF-α in vivo (10) suggests that inhibition of these cytokines by ruxolitinib and tofacitinib, which stimulates HIV-1 replication via induction of viral gene transcription (15, 19), could result in a decrease in viral replication......direct inhibition of proinflammatory cytokines by JAK inhibitors could, in turn, result in an overall downregulation of activation within cellular populations, thereby indirectly impacting autocrine and paracrine production of cytokines not directly controlled by the JAK-STAT pathway, such as TNF-α.....Other events could confer an overall reduced activation state and decreased permissiveness of HIV-1 target cells by inhibition of HIV-induced JAK-STAT pathway activation and indirect inhibition of paracrine and autocrine proinflammatory cytokine events.
HAART can achieve long-term viral suppression with certain limitations, including the inability to cure or address HIV-driven inflammation, necessitating the design of novel therapeutics with targets. It is well established that the JAK-STAT pathway is activated early in HIV-1 infection across multiple HIV-1 target cells including macrophages and lymphocytes (3, 4, 6, 9, 23), and this pathway orchestrates a multifaceted and tandem transduction of events, resulting in production of inflammatory factors, hyperactivation of the infected cell, IL-7 and IL-15 induction of homeostatic proliferation of central memory T cells (TCM) and transitional memory T cells (TTM) cells (30), which contributes to reservoir maintenance/reseeding, and global immune dysfunction across multiple sites including the gut, CNS, brain, and the systemic periphery (3, 4, 6, 9, 11-15, 17, 19, 23, 31). Activation of HIV-induced inflammation by induction of the JAK-STAT signaling cascade modulates multiple inflammation-driven pro-HIV events that favor virus replication, maintenance of viral reservoirs, and chronic infection across multiple sites systemically.
Selective and effective targeted inhibition of the JAK-STAT pathway could provide a multipronged mechanism to directly inhibit the JAK-STAT pathway, thereby inhibiting a pathway that modulates many pro-HIV events, and to significantly reduce systemic inflammation that drives disease progression and multiple pro-HIV events. In short, JAK1/2 inhibitors represent an attractive modality from which to confer indirect inhibition of HIV-1 replication by inhibiting a complex series of HIV-driven immunomodulatory events across lymphocytes and macrophages. JAK1/2 inhibitors were chosen for this study because JAK3 inhibitors present with off-target toxicity, including NK cell depletion (33), which can mitigate the ability of NK cells to modulate antiviral immunity, resulting in a transient increase in viral loads, as was observed in a recent study with SIV-infected rhesus macaques (33).
Relative to modulation of activation, as a function of inhibition of the JAK-STAT pathway, hyperactivation confers activation of the JAK-STAT pathway, and addition of a JAK inhibitor reduces this hyperactivation. As ruxolitinib and tofacitinib are well tolerated clinically and have approval for chronic long-term use in humans, it is evident that these drugs do not confer systemic and analogous inhibition of all JAK-STAT signaling, which would confer early significant toxicity. These factors imply that JAK inhibitors may selectively inhibit activation of cells that demonstrate hyperactivation of the JAK-STAT pathway, as would be the case for persons with myelofibrosis, rheumatoid arthritis, or, potentially, with HIV."
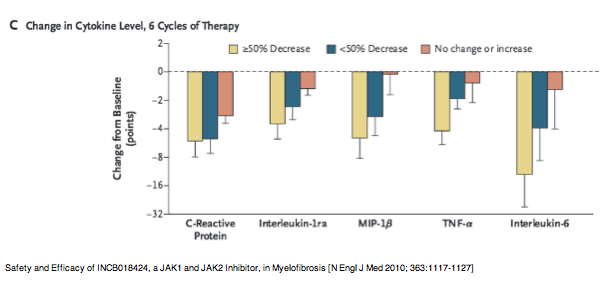
Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis
[N Engl J Med 2010; 363:1117-1127]
--------------
Ruxolitinib and Tofacitinib Are Potent and Selective Inhibitors of HIV-1 Replication and Virus Reactivation In Vitro
Antimicrobial Agents and Chemotherapy April 2014
Christina Gavegnano,a,b Mervi Detorio,a,b Catherine Montero,a,b Alberto Bosque,c Vicente Planelles,c Raymond F. Schinazia,b
Center for AIDS Research, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, Georgia, USAa; Veterans
Affairs Medical Center, Decatur, Georgia, USAb; Department of Pathology, University of Utah, Salt Lake City, Utah, USAc
ABSTRACT
The JAK-STAT pathway is activated in both macrophages and lymphocytes upon human immunodeficiency virus type 1 (HIV-1) infection and thus represents an attractive cellular target to achieve HIV suppression and reduced inflammation, which may impact virus sanctuaries. Ruxolitinib and tofacitinib are JAK1/2 inhibitors that are FDA approved for rheumatoid arthritis and myelofibrosis, respectively, but their therapeutic application for treatment of HIV infection was unexplored. Both drugs demonstrated submicromolar inhibition of infection with HIV-1, HIV-2, and a simian-human immunodeficiency virus, RT-SHIV, across primary human or rhesus macaque lymphocytes and macrophages, with no apparent significant cytotoxicity at 2 to 3 logs above the median effective antiviral concentration. Combination of tofacitinib and ruxolitinib increased the efficacy by 53- to 161-fold versus that observed for monotherapy, respectively, and each drug applied alone to primary human lymphocytes displayed similar efficacy against HIV-1 containing various polymerase substitutions. Both drugs inhibited virus replication in lymphocytes stimulated with phytohemagglutinin (PHA) plus interleukin-2 (IL-2), but not PHA alone, and inhibited reactivation of latent HIV-1 at low-micromolar concentrations across the J-Lat T cell latency model and in primary human central memory lymphocytes. Thus, targeted inhibition of JAK provided a selective, potent, and novel mechanism to inhibit HIV-1 replication in lymphocytes and macrophages, replication of drug-resistant HIV-1, and reactivation of latent HIV-1 and has the potential to reset the immunologic milieu in HIV-infected individuals.
INTRODUCTION
Although highly active antiretroviral agent therapy (HAART) can achieve long-term human immunodeficiency virus (HIV) suppression, current antiviral therapy does not achieve HIV eradication or a functional cure (1, 2). HAART has various shortcomings, including the inability to deliver adequate concentrations of drug to all HIV-1 target cells including macrophage-derived viral sanctuaries (1), rapid selection for emergence of drug-resistant variants/lack of efficacy against drug-resistant variants, lack of capacity to prevent reactivation of latent virus and subsequent systemic repopulation with virus, inability to mitigate HIV-orchestrated inflammation/immune dysfunction that drives infection and malignancies, inability to reduce or eliminate inflammation-driven HIV-associated neurocognitive impairments/activation of infected peripheral monocytes for trafficking to the brain/central nervous system (CNS), failure to prevent inflammation-driven priming of uninfected bystander cells for infection (1, 2), and a lack of impact on homeostatic proliferation of memory stem cells (Tscm). The inability to address all these factors necessitates the radical and innovative design of novel therapeutic treatments and modalities.
The Janus activating kinase-signal transducer and activator of transcription (JAK-STAT) pathway is activated early in HIV-1 infection across multiple HIV-1 target cells, including macrophages and lymphocytes (3, 4), and activation of this pathway orchestrates a multifaceted and tandem transduction of events resulting in production of inflammatory factors, hyperactivation of the infected cell, and global immune dysfunction across multiple sites including the CNS (5-7). Activation of HIV-induced inflammation by induction of the JAK-STAT signaling cascade modulates multiple pro-HIV events including the following: increased virus production in already infected cells, priming of uninfected bystander cells for infection, recruitment of uninfected cells to the site of infection, reactivation of virus from latent reservoirs, CNS infection/HIV-associated neurocognitive impairment, and promotion of HIV-orchestrated immune dysfunction in the gut and other organ sites (3-6, 8, 9). Therefore, potent, selective targeted inhibition of the JAK-STAT pathway could provide an attractive modality from which to confer indirect inhibition of HIV-1 replication by inhibiting a complex series of HIV-driven immunomodulatory events in various cells. It is possible that this will result in higher CD4+ counts, lower levels of immune activation and chronic inflammation, and improved event-free survival after a limited duration of JAK-STAT inhibitor treatment.
Two JAK1/2 inhibitors, ruxolitinib and tofacitinib, are FDA approved for myelofibrosis and rheumatoid arthritis, respectively. In humans, ruxolitinib inhibited various proinflammatory cytokines, including interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and IL-1 (10), and tofacitinib's approved use for rheumatoid arthritis underscores its potent in vivo anti-inflammatory effects. These cytokines are causative orchestrators of chronic inflammation, chronic infection, and disease progression (11-22), and together they may represent a significant obstacle that must be removed to achieve a functional cure or systemic eradication.
During our search of molecules that could interfere with pathways involved in promoting HIV replication, we discovered that certain JAK inhibitors selectively inhibited HIV in infected human and rhesus macaque primary lymphocytes and macrophages. Because these drugs interfere in vivo with the activation of the JAK-STAT pathway by HIV-1 infection (3, 4, 6, 9, 23, 24) and have specific anti-inflammatory properties, we further explored their potential as antiviral agents for HIV.
MATERIALS AND METHODS
Isolation and culture of lymphocytes and macrophages.For macrophage cultures, monocytes were isolated from buffy coats of HIV-1-negative, hepatitis B virus/hepatitis C virus (HBV/HCV)-negative donors with density gradient centrifugation coupled with enrichment for CD14+ monocytes with Rosette Sep antibody cocktail (Stem Cell Technologies, Vancouver, British Columbia). Wells were seeded at a density of 1 x 106 cells/well for 1 h at 37°C and 5% CO2 to confer plastic adherence prior to repeated washes with 1x phosphate-buffered saline (PBS). Macrophages were maintained in medium containing macrophage colony-stimulating factor (M-CSF) for 18 h prior to two washes with 1x PBS (to remove M-CSF) and subsequent culture in M-CSF-free medium supplemented with 20% heat-inactivated fetal calf serum and 1% penicillin-streptomycin for six more days prior to antiviral studies. For all conditions, macrophages were stained with CD11b-allophycocyanin (APC; Miltenyi Biotec, Auburn, CA) and subjected to fluorescence-activated cell sorting (FACS) to determine a purity of >99%.
Primary human peripheral blood mononuclear (PBM) cells were isolated from buffy coats derived from healthy donors (Lifesouth, Dunwoody, GA). Phytohemagglutinin (PHA)-stimulated PBM cells were maintained in RPMI medium (HyClone, Logan, UT) containing 6 μg/ml phytohemagglutinin (Cape Cod Associates, East Falmouth, MA) supplemented with 20% heat-inactivated fetal calf serum, 1% penicillin-streptomycin, and 2% l-glutamine (Sigma-Aldrich, San Jose, CA) for 72 h prior to proliferation/viability studies. PBM cells stimulated with PHA plus IL-2 were maintained analogously, with the exception of addition of human recombinant IL-2 (HR-IL-2; 26.5 units/ml) (Chiron Inc., Emeryville, CA) for 72 h prior to antiviral or proliferation/viability studies. Rhesus macaque lymphocytes or macrophages were isolated from whole blood obtained from the Yerkes National Primate Research Center (Atlanta, GA) and cultured as described above for human lymphocytes and macrophages. In addition, each animal was examined by the veterinarian monthly for weight, vital signs, physical exam (splenomegaly, lymph node enlargement, any signs of bruises, mouth exam, temperature, etc.), hemogram, hematocrit, etc., when blood samples were taken. Animals experiencing any wounds, infection, and/or diarrhea were given appropriate medication and care. The Yerkes Center is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), and all standard operating procedures are periodically reviewed by representatives from both the AAALAC and U.S. Department of Agriculture (USDA). The Yerkes Primate Research Center veterinary staff follows and/or exceeds the guidelines for the humane care of all animals at the Primate Center.
Antiviral studies.Antiviral potency is reported as the 50% effective concentration (EC50), or median effective concentration. Macrophages were cultured as described above for 7 days. For all antiviral studies, a multiplicity of infection (MOI) of 0.1 was used. Prior to infection, macrophages were serum starved for 8 h prior to infection and cultured for 2 h in medium with various concentrations of tofacitinib (Selleck Chemicals, Boston, MA, USA), ruxolitinib (Selleck Chemicals, Boston, MA, USA), or 3'-azido-3'-deoxythymidine (AZT; control) (ST Pharma Co., Ltd., Seoul, South Korea) prior to removal of drug-containing medium and 4-h infection with HIV-1BaL (human) or a simian-human immunodeficiency virus, RT-SHIV (rhesus macaque), at an MOI of 0.1 in the absence of drug. Then, virus was removed, and drug-containing medium was returned to each culture. RT-SHIV was chosen for macaque studies for the following reasons:(i) it allows flexibility of future combination studies using FDA-approved nonnucleoside reverse transcriptase inhibitors (NNRTI) as RT-SHIV contains a human reverse transcriptase, rendering this virus suitable for antiviral studies that may use NNRTI; (ii) it allows these data to be applicable for future studies in an RT-SHIV model wherein ruxolitinib and tofacitinib would be used with HAART; and (iii) it is otherwise analogous to simian immunodeficiency virus (SIV), rendering this virus suitable for comparison to traditional SIV infections as well. Supernatants were collected on day 7 postinfection, and HIV-1 p24 or p27 levels were measured by enzyme-linked immunosorbent assay (ELISA) (Zeptometrix Corp., Buffalo, NY). The median effective concentration (EC50)... and 90% effective concentration (EC90) were determined using CalcuSyn software (BioSoft Corporation, Cambridge, United Kingdom).
For lymphocytes, testing was performed using at least three independent assays performed in duplicate. Cells were incubated in RPMI medium (HyClone, Logan, UT) containing HR-IL-2 (26.5 units/ml) (Chiron, Inc., Emeryville, CA) and 20% heat-inactivated fetal calf serum. Infections were performed by adding HIV-1LAI or the molecular infectious cloned viruses containing the substitution K65R (HIV-1K65R), HIV-1M184V, HIV-1L74V, or HIV-1A62V/V75I/F77L/F116Y/Q151M or HIV-1 with four AZT-specific mutations [HIV-14AZT (D67N/K70R/T215Y/K219Q)], followed by a further incubation at 37°C in 5% CO2, 1 h prior to addition of drugs at 0.001, 0.01, 0.1, 1.0, 10, and 100 M. Assays were performed in 24-well plates (BD Biosciences, Franklin Lakes, NJ). One milliliter of supernatant was collected after 5 days in culture and then centrifuged at 12,000 rpm for 2 h at 4°C in a Jouan Br43i (Thermo Electron Corp., Marietta, OH). The product of the RT assay was quantified using a Packard harvester and direct beta counter, and the data were analyzed as previously described (25). Similar studies were performed using HIV-2pROD10 and HIV-1BAL (described above).
Cytotoxicity studies.Cytotoxicity of compounds is reported as 50% inhibitory concentration (IC50), or median inhibitory concentration. The IC50 was determined by propidium iodide staining (Invitrogen, Eugene, OR) according to the manufacturer's protocol (flow cytometry). For all assays, cells were cultured as described above and maintained in various concentrations of drug-containing medium for 6 days prior to assessment of toxicity. Cytotoxicity was considered when the concentrations of the test compounds alone inhibited growth by 50%.
For propidium iodide studies, PBM cells stimulated with PHA plus IL-2 were exposed to various concentrations of ruxolitinib or tofacitinib for 6 days prior to assessment of viability using propidium iodide (flow cytometry). Gating strategy based on forward scatter (FSC) and side scatter (SSC) was established and used uniformly across all samples (Fig. 1A). Cells incubated in the absence of drug were 92.8% viable, and cells exposed for 1 min at 95°C were 2.8% viable (Fig. 1B). Exposure to 95°C heat for 1 min is a positive control used for flow cytometry to ensure that dead cells scatter in the appropriate gate. Gating was established based on viable cells cultured in the absence of drug (Fig. 1B). An additional negative control, AZT, which is known to be nontoxic to lymphocytes, was incubated with cells at various concentrations and was not toxic at any concentration tested (Fig. 1C). As an additional positive control, cells exposed to various concentrations of cycloheximide demonstrated a dose-response toxicity (Fig. 1C). Gating was established based on viable cells cultured in the absence of drug (Fig. 1B).
Viability and proliferation assays in lymphocytes.Viability and proliferation (cell count) were measured using a Vi-Cell trypan blue system. Lymphocytes stimulated with PHA or PHA plus IL-2 were maintained as described above for 72 h prior to plating of cells in 24-well plates (BD Falcon, Franklin Lakes, NJ, USA) at a concentration of 1 x 106 cells/well in 1 ml of medium per well. Tofacitinib or ruxolitinib was maintained at various concentrations (0.1, 1.0, 10, or 100 μM) for 5 days in duplicate wells. Viability and total cell number were determined using the Vi-Cell system and reported as percent viable cells in each well and total cell number per well. Numbers were normalized to wells containing medium and cells without drug. Data reported were means and standard deviations for at least three independent experiments conducted with at least four pooled donors and duplicates within each experiment. Mean cell count and viability for cells maintained in drug-free medium are shown in Fig. 2 (dotted lines).
Combination studies assessing antiviral potency.To evaluate whether the combination of tofacitinib plus ruxolitinib was synergistic, additive, or antagonistic, drugs at the ratio of their respective EC50s, which was determined to be 1:4, were added at various concentrations to lymphocytes, and infections and antiviral potency were determined as described above. Drug interactions and combination indexes (CI) were analyzed using CalcuSyn (Biosoft, Ferguson, MO, USA), which allows automated simulation of synergism or antagonism. CI values of <1, 1, and >1 indicate synergism, additivity, and antagonism, respectively.
Reactivation of latent HIV-1 in primary human lymphocytes.Latently infected cultured central memory T (TCM) cells were prepared from primary naive cells, and reactivation of latent HIV-1 infection as previously described (26) was then triggered by antibody-mediated CD3/CD28 costimulation in the presence of 0.1, 1, 10, and 100 μM tofacitinib, ruxolitinib, or lamivudine (3TC; negative control). Production of HIV-1 was monitored by intracellular p24 by flow cytometry and compared to cells reactivated in the absence of drug.
Additionally, although the JAK-STAT pathway does not directly modulate TNF-α, JAK inhibitors exhibit potent inhibition of TNF-α in humans (10), providing a rationale for assessment of the ability of JAK inhibitors to inhibit TNF-α-triggered activation of HIV latency. The J-Lat latently infected T cell line was cultured as previously described (27) and then triggered to reactivate by 24 h of exposure to TNF-α (10 ng/ml) stimulation in the presence of 0.1, 1, 10, or 100 μM tofacitinib, ruxolitinib, or lamivudine (3TC). 3TC was chosen as a negative control because 3TC is a nucleoside reverse transcriptase inhibitor (NRTI) and therefore is not expected to impact green fluorescent protein (GFP) production in the J-Lat system, where there is no active reverse transcription. Reactivated virus was monitored by intracellular GFP production (flow cytometry).
Statistical methods.Means, standard deviations, and statistical comparisons using an unpaired Student t test were performed using the statistical routines in Microsoft Excel 2007. A P value of <0.05 was considered statistically significant.
RESULTS
Antiviral potency and cytotoxicity of ruxolitinib and tofacitinib in primary human lymphocytes and macrophages.The antiviral potencies of ruxolitinib and tofacitinib against HIV-1LAI in primary human lymphocytes were 0.1 to 0.8 μM (EC50) and 4.7 to 15.1 μM (EC90), respectively. The antiviral potencies of ruxolitinib and tofacitinib against HIV-2pROD10 in primary human lymphocytes were 0.02 to 0.07 μM (EC50) and 0.4 to 1.8 μM (EC90), respectively. Antiviral potencies of ruxolitinib and tofacitinib against HIV-1BaL in primary human macrophages were 0.3 μM (EC50) and 3.0 μM (EC90), respectively. As expected, AZT, used as a positive control, demonstrated high antiviral potency against HIV-1LAI, HIV-2, and HIV-1BaL. Cytotoxicity assays using propidium iodide (primary human lymphocytes) demonstrated no significant toxicity (IC50 of >50 μM). The therapeutic index (ratio of toxicity to potency) was >100 across both lymphocytes and macrophages for HIV-1 and HIV-2 (HIV-1/2) and RT-SHIV (Table 1).
Antiviral potency of ruxolitinib and tofacitinib in primary rhesus macaque lymphocytes and macrophages.Antiviral potencies were approximately 0.4 μM (EC50) and 4.0 μM (EC90) for both ruxolitinib and tofacitinib in activated primary rhesus macaque macrophages. Antiviral potencies for ruxolitinib and tofacitinib ranged from 0.09 to 0.3 μM (EC50) and from 1.3 to 2.9 μM (EC90) in primary rhesus macaque lymphocytes. As expected, AZT used as a positive control demonstrated selective antiviral potency. Data presented are the means and standard deviations from at least three independent experiments (Table 1).
Antiviral potency of ruxolitinib and tofacitinib against various NRTI-resistant HIV-1 strains in primary human lymphocytes.Efavirenz (EFV) was similarly potent across all NRTI-resistant strains as anticipated. Data presented are means and standard deviations calculated from at least four independent experiments, with pooled cells from eight donors and duplicates in each experiment. The antiviral potencies of ruxolitinib and tofacitinib were not markedly different for wild-type HIV-1xxLAI and HIV-1 containing substitutions K65R, M184V, L74V, A62V/V75I/F77L/F116Y/Q151M, or 4xAZT (D67N/K70R/T215Y/K219Q) (Table 2). The controls of AZT, emtricitabine [(-)-FTC], 3TC, stavudine (d4T), 2',3'-dideoxyinosine (ddI), efavirenz (EFV), and tenofovir disoproxil fumarate (TDF) demonstrated sensitivity or resistance as anticipated (Table 2). All data in Table 2 are reported as EC50/EC90 values.
Viability of primary human lymphocytes exposed to various concentrations of ruxolitinib or tofacitinib.Ruxolitinib did not significantly reduce viability versus no-drug controls for any concentration tested with the exception of 50 μM (P < 0.05) (Fig. 1D). Tofacitinib did not significantly reduce viability versus no-drug controls for any concentration tested (Fig. 1E). Histograms and scatter plots are representative data from at least three independent experiments conducted with pooled cells from eight donors. Cotreatment with ruxolitinib and tofacitinib at a ratio of 1:4 (lymphocytes) or 1:1 (macrophages) did not confer any significant increase in toxicity versus that observed for each drug administered alone (data not shown).
Effect of various JAK inhibitors on proliferation and viability of primary human lymphocytes stimulated with PHA or PHA plus IL-2.For PHA-stimulated lymphocytes, viability and proliferation were not significantly different from those of cells exposed to medium alone for all concentrations of either ruxolitinib or tofacitinib (Fig. 2A and C). Similarly, for lymphocytes stimulated with PHA plus IL-2, viability was not significantly different from that of cells exposed to medium alone for all concentrations of either ruxolitinib or tofacitinib (Fig. 2B); however, cell proliferation was inhibited by 1 μM ruxolitinib or tofacitinib (Fig. 2D).
Inhibition of reactivation of latent HIV-1 in lymphocytes by ruxolitinib and tofacitinib.Tofacitinib and ruxolitinib inhibited reactivation of latent HIV-1 in a primary central memory cell-based T cell latency model (Fig. 3A) and in the J-Lat latency T cell system, where the two JAK inhibitors were preincubated with cells for 30 min prior to addition of TNF-α (J-Lat) or CD3/CD28 (primary T cell model) in the presence of drug for 24 h to confer reactivation (Fig. 3B). Ruxolitinib was the more efficacious inhibitor across both systems and inhibited ≥50% of reactivation at steady-state concentrations or at the maximum concentration (Cmax) in vivo (Fig. 3, shaded boxes) (28, 29).
Synergistic antiviral potency for coadministration of ruxolitinib and tofacitinib in primary human lymphocytes and macrophages.For cotreatment with ruxolitinib and tofacitinib at a ratio of 1:4 (lymphocytes) or 1:1 (macrophages) (Fig. 4A and B, respectively), the EC50 decreased modestly by 2-fold for ruxolitinib and tofacitinib alone versus the combination. The EC90s decreased by 53-fold (tofacitinib) and 161-fold (ruxolitinib) (Fig. 4, dotted lines). The EC50 and EC90 were markedly decreased in macrophages (Fig. 4B). The CI value for the EC90 in lymphocytes was 0.07 ± 0.02, confirming synergism. Likewise, CI values for the EC50 and EC90 in macrophages were <0.1, confirming synergistic antiviral interactions.
DISCUSSION
HAART can achieve long-term viral suppression with certain limitations, including the inability to cure or address HIV-driven inflammation, necessitating the design of novel therapeutics with targets. It is well established that the JAK-STAT pathway is activated early in HIV-1 infection across multiple HIV-1 target cells including macrophages and lymphocytes (3, 4, 6, 9, 23), and this pathway orchestrates a multifaceted and tandem transduction of events, resulting in production of inflammatory factors, hyperactivation of the infected cell, IL-7 and IL-15 induction of homeostatic proliferation of central memory T cells (TCM) and transitional memory T cells (TTM) cells (30), which contributes to reservoir maintenance/reseeding, and global immune dysfunction across multiple sites including the gut, CNS, brain, and the systemic periphery (3, 4, 6, 9, 11-15, 17, 19, 23, 31). Activation of HIV-induced inflammation by induction of the JAK-STAT signaling cascade modulates multiple inflammation-driven pro-HIV events that favor virus replication, maintenance of viral reservoirs, and chronic infection across multiple sites systemically.
Specific cytokines, including IL-6, TNF-α, and IL-1α/ß, have been implicated as key factors in disease progression and maintenance of replication and infection (11-13, 15, 19, 22). Levels of circulating IL-6 correlate with residual viremia and are a marker for immune dysfunction and microbial translocation (18, 21). Additionally, multiple reports demonstrate that IL-6 levels remain elevated in patients with well-controlled viremia (15, 18, 31) and correlate this elevation to persistent low-level viremia that may seed reservoirs and contribute to an inability to eradicate virus even when the majority of replication is controlled. Other reports confirm that immune activation is a strong predictor for disease progression (17, 20, 21, 32) and state that immune activation at treatment baseline determines the extent of immune reconstitution that is possible thereafter (16), underscoring the importance of controlling inflammation across all individuals, independent of viral load or disease state. In keeping with the established role of inflammation in HIV persistence, many reports correlate activation of the JAK-STAT pathway with multiple pro-HIV events, including apoptosis of monocytes in HIV-infected persons (23), continued HIV-orchestrated increase in inflammatory responses that drive migration of infected cells across the blood-brain barrier (BBB) (6), and JAK-STAT-mediated perpetuation of a proinflammatory phenotype in myeloid lineage cells (9).
Selective and effective targeted inhibition of the JAK-STAT pathway could provide a multipronged mechanism to directly inhibit the JAK-STAT pathway, thereby inhibiting a pathway that modulates many pro-HIV events, and to significantly reduce systemic inflammation that drives disease progression and multiple pro-HIV events. In short, JAK1/2 inhibitors represent an attractive modality from which to confer indirect inhibition of HIV-1 replication by inhibiting a complex series of HIV-driven immunomodulatory events across lymphocytes and macrophages. JAK1/2 inhibitors were chosen for this study because JAK3 inhibitors present with off-target toxicity, including NK cell depletion (33), which can mitigate the ability of NK cells to modulate antiviral immunity, resulting in a transient increase in viral loads, as was observed in a recent study with SIV-infected rhesus macaques (33). Interestingly, this report describes a lack of decline in CD4 T cells despite transient increases in viral loads. This sustained CD4 T cell count in the presence of higher viral loads is likely conferred by the antiviral and anti-inflammatory effects of the JAK inhibitor that was administered. Use of a JAK1/2 inhibitor could confer antiviral and anti-inflammatory benefits without the off-target toxicity of NK cell depletion that appear with a JAK3 inhibitor.
Herein, we report for the first time that two FDA-approved JAK inhibitors, ruxolitinib and tofacitinib, are effective, submicromolar inhibitors of HIV replication in lymphocytes and macrophages. Infections were performed as acute infection, where uninfected cells were exposed to drug prior to infection with HIV-1, and viral quantification was performed at 5 to 7 days postinfection. This experimental design allows for assessment of the impact of tofacitinib and ruxolitinib on establishment of the first round of viral replication in uninfected cells, as well as multiple rounds of infection thereafter.
Additionally, as data are collected from uninfected cells exposed to virus for 5 to 7 days, the reduction in virus observed at this late time point, which is a function of both inhibition of de novo infection and subsequent spread to new cells, implies that inhibition of viral replication is transpiring. Determination of the impact of JAK inhibitors on other parameters such as the reduction of integrated viral DNA, size, and half-life of viral reservoirs will be reported elsewhere.
Complementary to these data, both inhibitors demonstrated similar potencies against HIV-2 infection in human lymphocytes and against RT-SHIV infection in rhesus macaque lymphocytes and macrophages (Table 1). These data were obtained with PBM cells stimulated with PHA and IL-2 prior to infection to facilitate a tandem activation of the cells by both PHA (a nonspecific mechanism) and IL-2 (specific mechanism) to ensure that cells are activated, proliferating, and therefore permissive to HIV-1, HIV-2, or SHIV infection. The combination of PHA plus IL-2 is traditionally used to prime primary PBM cells for infection and was therefore also employed for these assays. Additionally, use of a mitogen, to confer early nonspecific activation, in tandem with IL-2, to confer specific, receptor-mediated activation and proliferation, is intended to create an environment that is similar to that observed in vivo, where the presence of virus in an infected individual provokes a chronic state of activation and priming of uninfected cells for infection. Ruxolitinib at clinically approved doses demonstrated a greater than 50% reduction in IL-6, TNF-α, and IL-1 receptor antagonist (IL-1Ra) (10), and the primary indication for tofacitinib use is rheumatoid arthritis, indicating its strong anti-inflammatory capacity in vivo. As the combination of PHA plus IL-2 was used as a mitogen prior to infection and as it induces a hyperactivated, proliferating state in the cell population in tandem with HIV-1, HIV-2, or RT-SHIV infection, it is plausible that the antiviral potency observed with ruxolitinib and tofacitinib is a function of modulation of proinflammatory cytokines, which in turn alters the activation state of the PBM cells, conferring a downregulated activation state. The established inhibition of inflammation by these drugs provides a foundation for an indirect inhibition of the viral replication cycle by interference with inflammatory factors that promote productive viral replication. The antiviral potency observed with ruxolitinib and tofacitinib in macrophages suggests that effective inhibition of viral replication across sentinel myeloid-derived viral reservoirs is possible in vivo, underscoring the ability of these inhibitors to address ongoing replication across cellular compartments, where current antiretroviral therapy is delivered at subtherapeutic concentrations (1).
Activation of STATs by the JAK-STAT pathway is associated with increased inflammation and leukocyte transmigration across the BBB (6), underscoring the sentinel role of JAK-STAT activation in inflammation and immune activation, which governs systemic pro-HIV events. In addition, the anti-inflammatory profile of these compounds could mitigate trafficking of infected, activated CD14+/CD16+ monocytes in the periphery across the BBB and into the CNS (6, 7, 12, 13). This JAK inhibitor-mediated blockade could significantly reduce HIV-associated neurocognitive dysfunction, which is often conferred by inflammation-driven events within the CNS compartments (6, 7, 12, 13, 17, 22).
The established anti-inflammatory profile of ruxolitinib and tofacitinib suggests that these compounds may be effective inhibitors of homeostatic proliferation and reservoir maintenance of TCM, TTM, or TSCM cells, which is driven by IL-7 and IL-15, two proinflammatory cytokines (30, 34). Concurrently, it is possible that a reduction in systemic inflammation and activation could lead to reduction of HIV-1 coreceptor expression including CCR5 as its expression is positively correlated with activation state (35, 36). This mechanism could reduce HIV-mediated entry into permissive cells through an indirect, activation state-driven reduction in CCR5 expression. It follows that decreased cellular activation could result in fewer de novo cell-to-cell HIV transmission events, which would have otherwise occurred through ART-independent transmission of virus from activated T cells. This suggests that JAK inhibitors would thereby confer fewer latency-establishing events. The ability of ruxolitinib and tofacitinib to inhibit IL-6 and TNF-α in vivo (10) suggests that inhibition of these cytokines by ruxolitinib and tofacitinib, which stimulates HIV-1 replication via induction of viral gene transcription (15, 19), could result in a decrease in viral replication. Although the JAK-STAT pathway is not directly activated by TNF-α, in vivo inhibition of JAK-STAT signaling by ruxolitinib significantly decreases peripheral TNF-α levels (10), clearly defining a link between ruxolitinib administration and TNF-α levels.
Additionally, direct inhibition of proinflammatory cytokines by JAK inhibitors could, in turn, result in an overall downregulation of activation within cellular populations, thereby indirectly impacting autocrine and paracrine production of cytokines not directly controlled by the JAK-STAT pathway, such as TNF-α. Together, these multifaceted and independent mechanisms could result in a tandem and simultaneous anti-HIV effect. Some events may directly inhibit viral gene transcription by inhibiting IL-6 and TNF-α, which are associated with induction of viral gene transcription (15, 19). Other events could confer an overall reduced activation state and decreased permissiveness of HIV-1 target cells by inhibition of HIV-induced JAK-STAT pathway activation and indirect inhibition of paracrine and autocrine proinflammatory cytokine events.
The degree to which ruxolitinib and tofacitinib inhibit JAK1, JAK2, JAK3, or Tyk2 and the downstream effects of this inhibition can be significantly altered as a function of differential JAK inhibition. Therefore, it follows that two different inhibitors, although from the same drug class, could confer synergistic antiviral effects due to differences relative to JAK inhibition profiles. Thus, coadministration of ruxolitinib and tofacitinib at ratios of their respective EC50 values (1:1 in macrophages and 1:4 in lymphocytes) demonstrated synergistic antiviral potency in both lymphocytes and macrophages and significantly decreased the concentration of each drug required to inhibit viral replication (Fig. 4). Although the exact mechanism for synergism is not defined, it follows that differential inhibition of JAK1, JAK2, JAK3, and Tyk2 by each drug creates multiple targets within the JAK-STAT pathway from which to confer inhibition of proinflammatory cytokines, thereby making it possible for synergism to occur.
Relative to modulation of activation, as a function of inhibition of the JAK-STAT pathway, hyperactivation confers activation of the JAK-STAT pathway, and addition of a JAK inhibitor reduces this hyperactivation. As ruxolitinib and tofacitinib are well tolerated clinically and have approval for chronic long-term use in humans, it is evident that these drugs do not confer systemic and analogous inhibition of all JAK-STAT signaling, which would confer early significant toxicity. These factors imply that JAK inhibitors may selectively inhibit activation of cells that demonstrate hyperactivation of the JAK-STAT pathway, as would be the case for persons with myelofibrosis, rheumatoid arthritis, or, potentially, with HIV. To test this hypothesis, we evaluated the ability of ruxolitinib and tofacitinib to inhibit proliferation of primary human lymphocytes stimulated with PHA or PHA plus IL-2. Addition of ruxolitinib or tofacitinib did not reduce viability across either cell activation state for all concentrations tested (Fig. 2A and B) and did not alter PHA-stimulated proliferation (Fig. 2C). Both drugs demonstrated effective inhibition of proliferation stimulated by PHA plus IL-2 but not solely by PHA in a dose-dependent manner (Fig. 2D), implying that ruxolitinib and tofacitinib selectively inhibit proliferation in hyperactivated populations of cells. All viability/proliferation studies were conducted with human PBM cells exposed to either PHA or PHA plus IL-2 for 6 days. In contrast, all antiviral studies were conducted with PHA plus IL-2, and ruxolitinib and tofacitinib inhibited viral replication in these cells. The time point of 5 days was chosen to assess the impact of ruxolitinib and tofacitinib under in vitro conditions similar to those which may occur in vivo, where activation is conferred during earlier time points by initial cytokine-mediated activation but where subsequent paracrine and autocrine events will transpire after the initial peak of activation and will persist thereafter. As it is well documented that the JAK-STAT pathway is activated in HIV-infected cells (3, 4, 6, 8, 23), it follows that the antiviral mechanism of action of ruxolitinib and tofacitinib could, in part, be related to specific inhibition of JAK-STAT-mediated signaling in HIV-infected cells, which harbor higher levels of JAK-STAT activation than uninfected cells (3, 4, 9). Additionally, reports have demonstrated that various STAT binding sites exist in the long terminal repeats of various HIV-1 subtypes (37) and that cytokine-induced activation of STAT5 increases HIV-1 production in primary CD4+ T cells (24). These findings give credence to the hypothesis that ruxolitinib- and tofacitinib-mediated blockade of STAT activation confers antiviral potency, independent of whether cell-specific STAT activation is conferred by pro-HIV cytokines or directly by HIV-1/2. These findings also provide a foundation for a hypothesis framed upon the potential for a cellular-factor-based inhibition of viral replication by JAK inhibitors. As we have not been able to select for resistant virus even after 18 weekly passages, cotreatment of infected cells with a JAK inhibitor plus traditional HAART could increase the amount of time for traditional ART resistance to develop or potentially modulate the resistance pattern that emerges.
Interestingly, the concentrations of ruxolitinib or tofacitinib that conferred inhibition of reactivation, for both TNF-α- and CD3/CD28-mediated reactivation, are 1 to 2 logs greater than those which confer antiviral potency across all cell types tested, demonstrating that the mechanism that confers inhibition of reactivation may be independent of that which confers antiviral activity.
Although the JAK-STAT pathway is not directly activated by TNF-α, a TNF-α-based latency model was specifically chosen for the following reasons: (i) in vivo inhibition of JAK-STAT signaling by ruxolitinib significantly decreases peripheral TNF-α levels (10), clearly defining a link between ruxolitinib administration and TNF-α; (ii) direct inhibition of proinflammatory cytokines by JAK inhibitors could, in turn, result in an overall downregulation of activation within cellular populations, thereby indirectly impacting autocrine and paracrine production of cytokines not directly controlled by the JAK-STAT pathway, such as TNF-α; and (iii) the direct mechanism by which ruxolitinib confers a significant in vivo reduction in TNF-α is unknown, and the mechanism(s) responsible for this cross talk could also modulate reactivation of latent HIV-1, which is also regulated by complex cross talk involving cytokines and cellular signaling events.
Together, these data demonstrate that targeted inhibition of the JAK-STAT pathway by inhibition of JAK provides a selective, effective, and novel mechanism to inhibit HIV-1 replication in lymphocytes and macrophages, replication of drug-resistant HIV-1, and reactivation of latent HIV-1. Inhibition of the JAK-STAT pathway using FDA-approved agents, such as ruxolitinib and tofacitinib, provides the first opportunity to have a significant impact on this inflammation and may reset the immunologic milieu.
These findings present a compelling rationale for further work designed to elucidate the potential clinical relevance of these drugs in a macaque model and eventually in humans at a safe and effective dose.
|
|
| |
| |
|
|
|