 |
|
|
| |
Emerging Treatments for ASH & NASH - Rohit Loomba, MD / AASLD 2016
|
| |
| |
Download the PDF here
from a talk at AASLD 2016 by Rohit Loomba MD

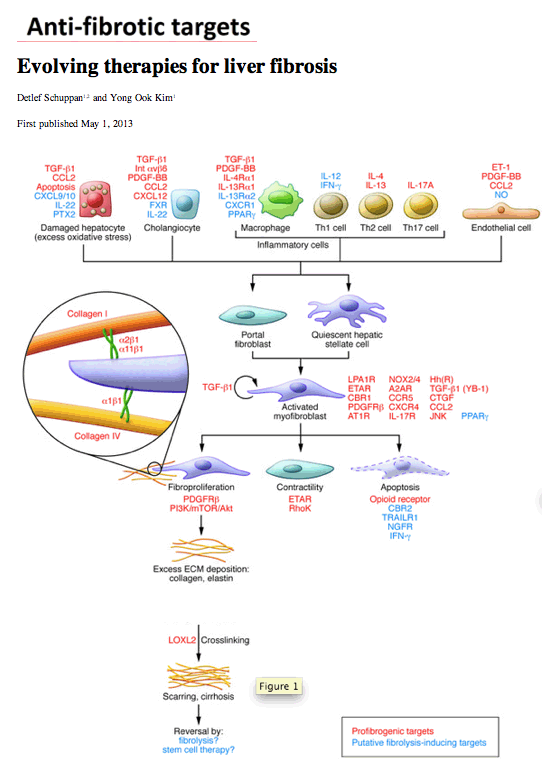
Figure 1
Myofibroblasts and their fibrogenic activation.
Cells and major factors upstream of quiescent portal fibroblasts and hepatic stellate cells that induce transformation to fibrogenic myofibroblasts. This schematic highlights several major targets to treat liver fibrosis. Notably, the ECM itself can serve as modulator of fibrogenesis and fibrolysis. Thus collagen fibrils become crosslinked by LOXL2, which contributes to the reduced reversibility of advanced fibrosis, and collagen-binding ECM receptors (especially the integrins α1β1, α2β1, and α11β1) confer signals of stress or stress relaxation that either maintain fibrogenic activation or induce fibrolytic activity of the myofibroblasts. Additional minor contributors to fibrogenic activation are not shown here (see text for details). A2AR, adenosine 2A receptor; AT1R, angiotensin 1 receptor; CBR1, cannabinoid receptor 1; ET-1, endothelin-1; ETAR, endothelin A receptor; FXR, farnesoid X receptor; Hh(R), hedgehog (receptor); Int, integrin; LPA1R, lysophosphatidic acid receptor 1; NGFR, nerve growth factor receptor; PTX2, pentraxin 2; TRAILR, TNF-related apoptosis-inducing ligand receptor; YB-1, Y-box binding protein.
Fibrosis is the excess accumulation of ECM, which results from chronic, nonresolving inflammation. This inflammation triggers a wound-healing process that mitigates inflammatory tissue destruction but also leads to scar tissue formation. In the liver, fibrosis is mainly due to chronic viral hepatitis B or C, autoimmune and biliary diseases, alcoholic steatohepatitis (ASH) and, increasingly, nonalcoholic steatohepatitis (NASH) (1-5). While mild fibrosis remains largely asymptomatic, its progression toward cirrhosis, i.e., replacement of functional parenchyma by scar tissue accompanied by severe architectural and vascular distortion, is the major cause of liver-related morbidity and mortality. Clinical sequelae of cirrhosis are (a) liver synthetic (functional) failure, including failing hemostatic, nitrogen handling, and detoxification systems; (b) portal hypertension with consequent formation of ascites and bleeding esophageal or gastric varices; (c) a high susceptibility to infection; and (d) a high risk to develop hepatocellular carcinoma (HCC) (2). Preventive measures, such as antiviral regimens for hepatitis B or C, are already decreasing the burden of viral cirrhosis and HCC, but other causes, such as NASH (which is linked to obesity and type 2 diabetes) are taking center stage. Moreover, numerous patients present initially in the clinic with advanced fibrosis or cirrhosis, which are largely irreversible. Therefore, antifibrotics that prevent progression toward cirrhosis or induce regression of advanced fibrosis and cirrhosis are urgently needed (6-9).
Research has delineated key mechanisms and cells that determine fibrosis progression (fibrogenesis) and regression (fibrolysis) (1-19). Notably, liver fibrosis has much in common with fibrosis of other organs, such as lungs and kidneys, leading to a cross-fertilization of research across organ boundaries. The structural components of the fibrotic ECM, the growth factors, cytokines, chemokines, and proteases, as well as central signaling cascades implicated in fibrogenesis and fibrolysis, are nearly identical in these different tissues (18, 20-22). Importantly, fibrosis is no longer considered static, but the result of a continuous remodeling process. Nonetheless, in contrast to kidneys and lungs, the liver has an extraordinary capacity to regenerate, even in advanced fibrosis.
Fibrosis is intimately linked to wound healing, serving to prevent tissues from disassembly during inflammation, apoptosis, necrosis, and release of lytic enzymes.
Fibrosis usually reverses within days to a few weeks following the resolution of tissue damage, as demonstrated in less advanced rodent and human liver fibrosis (2, 8, 9, 23-25). However, the longer the damage persists, often at a low level, the more ECM is deposited. This chronic damage results in increasingly acellular scar tissue and a steep decline of potential reversibility, even after elimination of causative triggers (26, 27).
Inefficient fibrolysis is due to several factors: (a) lack of cues for ordered cell repopulation and regeneration due to an atypical ECM and the loss of appropriate cellular context, (b) advanced vascular remodeling with architectural distortion, (c) extensive crosslinking of ECM components such as fibrillar collagen that make proteolytic removal difficult, and (d) the disappearance of cellular elements that digest the scar tissue. Here we discuss the cellular and molecular pathways that promote fibrosis progression and highlight current clinical trials as well as improved methods of monitoring fibrosis.
Research has delineated key mechanisms and cells that determine fibrosis progression (fibrogenesis) and regression (fibrolysis) (1-19). Notably, liver fibrosis has much in common with fibrosis of other organs, such as lungs and kidneys, leading to a cross-fertilization of research across organ boundaries. The structural components of the fibrotic ECM, the growth factors, cytokines, chemokines, and proteases, as well as central signaling cascades implicated in fibrogenesis and fibrolysis, are nearly identical in these different tissues (18, 20-22). Importantly, fibrosis is no longer considered static, but the result of a continuous remodeling process. Nonetheless, in contrast to kidneys and lungs, the liver has an extraordinary capacity to regenerate, even in advanced fibrosis.
Fibrosis is intimately linked to wound healing, serving to prevent tissues from disassembly during inflammation, apoptosis, necrosis, and release of lytic enzymes. Fibrosis usually reverses within days to a few weeks following the resolution of tissue damage, as demonstrated in less advanced rodent and human liver fibrosis (2, 8, 9, 23-25). However, the longer the damage persists, often at a low level, the more ECM is deposited. This chronic damage results in increasingly acellular scar tissue and a steep decline of potential reversibility, even after elimination of causative triggers (26, 27).
Inefficient fibrolysis is due to several factors: (a) lack of cues for ordered cell repopulation and regeneration due to an atypical ECM and the loss of appropriate cellular context, (b) advanced vascular remodeling with architectural distortion, (c) extensive crosslinking of ECM components such as fibrillar collagen that make proteolytic removal difficult, and (d) the disappearance of cellular elements that digest the scar tissue. Here we discuss the cellular and molecular pathways that promote fibrosis progression and highlight current clinical trials as well as improved methods of monitoring fibrosis.
|
| |
|
|
|
|
|