| |
|
Glecaprevir and Pibrentasvir in Patients with HCV and Severe Renal Impairment
|
| |
| |
Download the PDF here
Download the PDF here
NEJM Oct 12 2017 - Edward Gane, M.D., Eric Lawitz, M.D., David Pugatch, M.D., Georgios Papatheodoridis, M.D., Norbert Bräu, M.D., Ashley Brown, M.D., Stanislas Pol, M.D., Ph.D., Vincent Leroy, M.D., Ph.D., Marcello Persico, M.D., Christophe Moreno, M.D., Ph.D., Massimo Colombo, M.D., Eric M. Yoshida, M.D.,
David R. Nelson, M.D., Christine Collins, Ph.D., Yang Lei, Ph.D., Matthew Kosloski, Ph.D., and Federico J. Mensa, M.D.
Abstract
Background
Chronic hepatitis C virus (HCV) infection is more prevalent among patients who have chronic kidney disease than among those who do not have the disease. Patients with chronic kidney disease who also have HCV infection are at higher risk for progression to end-stage renal disease than those who have chronic kidney disease without HCV infection. Patients with both HCV infection and advanced chronic kidney disease have limited treatment options.
Methods
We conducted a multicenter, open-label, phase 3 trial to evaluate the efficacy and safety of treatment with the combination of the NS3/4A protease inhibitor glecaprevir and the NS5A inhibitor pibrentasvir for 12 weeks in adults who had HCV genotype 1, 2, 3, 4, 5, or 6 infection and also had compensated liver disease (with or without cirrhosis) with severe renal impairment, dependence on dialysis, or both. Patients had stage 4 or 5 chronic kidney disease and either had received no previous treatment for HCV infection or had received previous treatment with interferon or pegylated interferon, ribavirin, sofosbuvir, or a combination of these medications. The primary end point was a sustained virologic response 12 weeks after the end of treatment.
Results
Among the 104 patients enrolled in the trial, 52% had genotype 1 infection, 16% had genotype 2 infection, 11% had genotype 3 infection, 19% had genotype 4 infection, and 2% had genotype 5 or 6 infection. The sustained virologic response rate was 98% (102 of 104 patients; 95% confidence interval, 95 to 100). No patients had virologic failure during treatment, and no patients had a virologic relapse after the end of treatment. Adverse events that were reported in at least 10% of the patients were pruritus, fatigue, and nausea. Serious adverse events were reported in 24% of the patients. Four patients discontinued the trial treatment prematurely because of adverse events; three of these patients had a sustained virologic response.
Conclusions
Treatment with glecaprevir and pibrentasvir for 12 weeks resulted in a high rate of sustained virologic response in patients with stage 4 or 5 chronic kidney disease and HCV infection. (Funded by AbbVie; ClinicalTrials.gov number, NCT02651194.)
Hepatitis C virus (HCV) infection is more prevalent among patients with chronic kidney disease than among those without the disease.1-3 Patients who have chronic kidney disease and concomitant HCV infection are also at higher risk for progression to end-stage renal disease,4,5 as well as for compensated cirrhosis and hepatocellular carcinoma,6-8 than patients who have chronic kidney disease without HCV infection. Renal insufficiency is an important extrahepatic manifestation of HCV infection.9 The development of HCV-related cirrhosis and consequent portal hypertension can complicate potential kidney transplantation for these patients. Furthermore, patients with HCV infection who undergo long-term hemodialysis pose a high risk of HCV transmission in dialysis centers, making HCV treatment of high importance for this population.
For patients with severe renal impairment and HCV genotype 2, 3, 5, or 6 infection, no all-oral, direct-acting antiviral regimens have been approved by any agency to date; the only currently approved treatment regimen is interferon with ribavirin. However, the negative side-effect profile of interferon is well documented in this population,10 and ribavirin is excreted in the urine, accumulates systemically in patients with severe renal impairment, and is associated with adverse events, such as hemolytic anemia and pruritus, thus exacerbating adverse events in a population that is already at high risk for anemia and cardiovascular events.11,12 Many direct-acting antiviral agents, including those coformulated with the nucleotide polymerase inhibitor sofosbuvir, are not recommended in patients with severe renal insufficiency since clearance of those agents occurs primarily in the kidneys.6,13-16 The direct-acting antiviral combination of elbasvir and grazoprevir, administered for 12 weeks, is a recommended ribavirin-free treatment option for patients with end-stage renal disease and HCV genotype 1 or 4 infection17,18; after 12 weeks of treatment, the regimen resulted in a sustained virologic response rate of 94%.19 For patients with HCV genotype 1a or 1b infection, 12 weeks of treatment with ombitasvir-paritaprevir-ritonavir and dasabuvir is another recommended treatment option that resulted in a high sustained virologic response rate at 12 weeks in patients with advanced renal disease20,21; however, for patients with HCV genotype 1a infection, the product label recommends coadministration with ribavirin. Overall, treatment options for many patients with stage 4 or 5 chronic kidney disease and HCV infection are limited, and to date, no treatment options are available that are free of both interferon and ribavirin and are universally recommended for patients with HCV genotype 2, 3, 5, or 6 infection.
Glecaprevir, an NS3/4A protease inhibitor, and pibrentasvir, an NS5A inhibitor, exhibit potent antiviral activity across all six major HCV genotypes.22,23 Phase 1 studies have shown that the metabolism and clearance of both glecaprevir and pibrentasvir occur primarily in the biliary system and that renal excretion of each of the two medications is negligible; consequently, dose adjustment is typically not needed for patients with severe renal impairment.24 In addition, a pharmacokinetic study showed that exposure levels of glecaprevir and pibrentasvir were not affected by hemodialysis; changes in exposure during dialysis for each compound were considered to be clinically insignificant (<7% for glecaprevir and <18% for pibrentasvir).25
In the phase 3 trial EXPEDITION-4, we assessed the efficacy and safety of ribavirin-free, coformulated glecaprevir (NS3/4A protease inhibitor) and pibrentasvir (NS5A inhibitor; formerly ABT-530) administered for 12 weeks in patients who had chronic HCV genotype 1, 2, 3, 4, 5, or 6 infection and also had stage 4 or 5 chronic kidney disease. In addition, patients could have had compensated cirrhosis, could have received previous treatment for HCV infection, or both. We also assessed patient-reported outcomes, but those results are not reported here.
Methods
Trial Design and Oversight
In this multicenter, open-label, single-group, phase 3 trial, patients received three coformulated tablets, each containing glecaprevir (100 mg) (AbbVie and Enanta Pharmaceuticals) and pibrentasvir (40 mg) (AbbVie) for a total dose of 300 mg of glecaprevir and 120 mg of pibrentasvir, once daily for 12 weeks. An overview of the trial design is provided in Figure S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. All the patients provided written informed consent, and the trial was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and the ethical principles of the Declaration of Helsinki. Personnel from AbbVie (the sponsor of the trial) contributed to the design of the trial and to the collection, analysis, and interpretation of the data and participated in the writing, review, and approval of the content of the manuscript. All the authors had access to the relevant data. The authors vouch for the completeness and accuracy of the data and for the fidelity of the trial to the protocol, which is available at NEJM.org.
Patient Population
Patients were screened between December 21, 2015, and March 25, 2016, at 30 trial centers in Australia, Belgium, Canada, France, Greece, Italy, New Zealand, the United Kingdom, and the United States; the first patient was enrolled on January 11, 2016. We enrolled adults 18 years of age or older who had chronic HCV genotype 1, 2, 3, 4, 5, or 6 infection and compensated liver disease with or without cirrhosis. Patients were required to have an estimated glomerular filtration rate at screening of less than 30 ml per minute per 1.73 m2 of body-surface area. Patients could have received either no previous HCV treatment or previous treatment with any combination of interferon or pegylated interferon, ribavirin, or sofosbuvir. Patients who had both HCV genotype 3 infection and previous treatment for HCV were excluded, since the most effective treatment regimen for such patients had not yet been established at the time of initiation of the current trial. Complete patient eligibility criteria are provided in the Supplementary Appendix. A diagram of patient screening and enrollment is provided in Figure S2 in the Supplementary Appendix.
Assessments and End Points
Efficacy End Points
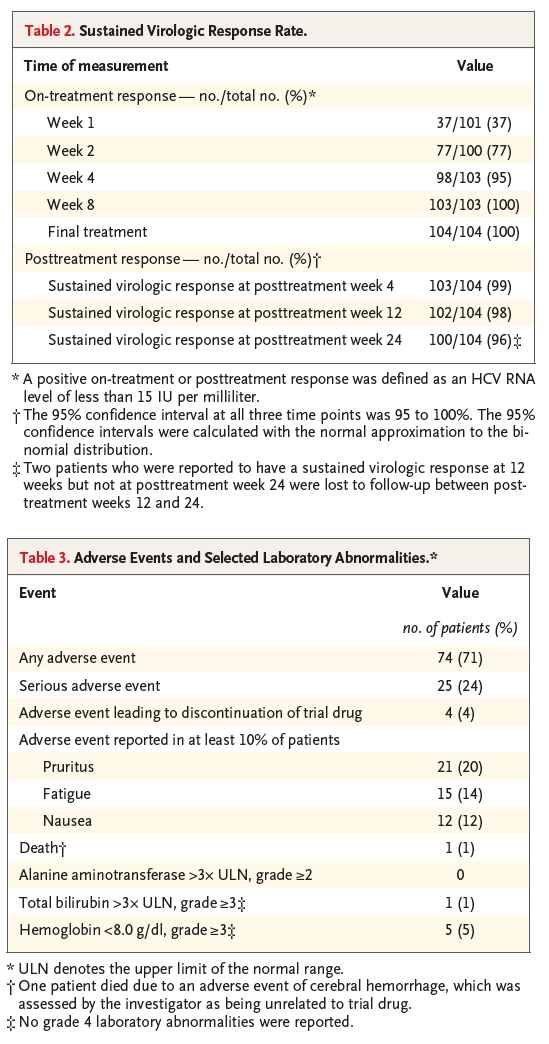
The primary efficacy end point of the trial was a sustained virologic response at 12 weeks, defined as an HCV RNA level of less than 15 IU per milliliter 12 weeks after the end of treatment. The secondary end points were the percentage of patients who had virologic failure during treatment and the percentage of patients who had a virologic relapse after treatment. Any patient who met one of the following criteria was considered to have had on-treatment virologic failure and was required to discontinue treatment: an increase in the HCV RNA level of at least 100 IU per milliliter after a measurement showing an HCV RNA level of less than 15 IU per milliliter during treatment, a confirmed increase in the HCV RNA level of more than 1 log10 IU per milliliter from the nadir during the treatment period, or an HCV RNA level of at least 15 IU per milliliter after 6 weeks of treatment. Patients who completed treatment and had an HCV RNA level of less than 15 IU per milliliter at the end of treatment were considered to have had a virologic relapse if they had a confirmed HCV RNA level of at least 15 IU per milliliter between the end of treatment and 12 weeks after the last dose of the trial drug. Additional prespecified efficacy end points included the percentage of patients who had a sustained virologic response 4 weeks and 24 weeks after the end of treatment.
Efficacy and Safety Assessments
Plasma samples were obtained at each treatment visit (weeks 1, 2, 4, 8, and 12) and at each posttreatment visit (posttreatment weeks 2, 4, 8, 12, and 24) for the assessment of HCV RNA levels. Plasma HCV RNA levels were measured with the use of a real-time reverse-transcriptase-polymerase-chain-reaction assay (COBAS AmpliPrep/COBAS TaqMan HCV Test, version 2.0, Roche). The lower limits of both detection and quantification were 15 IU per milliliter, irrespective of HCV genotype.
Safety assessments included the evaluation of adverse events, vital signs, physical examinations, electrocardiography, and laboratory testing. For all the patients who received at least one dose of the trial drug, adverse events were monitored during the treatment period and for 4 weeks after treatment completion; other safety end points were also measured during the treatment and posttreatment periods. Serious and nonserious adverse events were recorded from the date that written informed consent was provided through 30 days after discontinuation of the trial drug. Adverse events, including certain laboratory abnormalities that were recorded as adverse events, were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Relatedness of adverse events to the trial drug was determined by the investigator, who was unaware of the treatment assignments.
Virologic Resistance
We determined the presence of resistance-associated polymorphisms at baseline by performing next-generation sequencing on plasma samples obtained from each patient on day 1. Among the patients who did not have a sustained virologic response at 12 weeks, full-length sequencing of NS3/4A and NS5A genes was performed on all available plasma samples with an HCV RNA level of at least 1000 IU per milliliter. Virologic amino acid variants were identified by comparison with the appropriate prototypical reference sequence, at a population detection threshold of 15%.
Statistical Analysis
We planned to enroll approximately 100 patients and specified no formal statistical hypothesis to be tested. Analysis of the primary end point was conducted after all the enrolled patients had completed the posttreatment week 12 visit. Efficacy and safety analyses were based on the intention-to-treat population, which comprised all patients who received at least one dose of the trial drug. We determined the percentage of patients who met the criteria for each of the primary and secondary end points of the trial, and we calculated a two-sided 95% confidence interval using the normal approximation to the binomial distribution.
Results
Patients
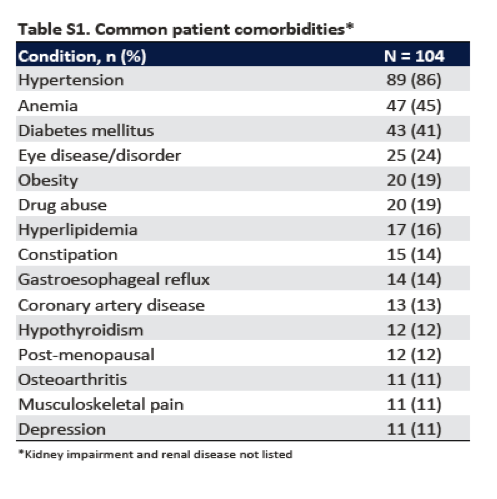
The demographic, disease, and clinical characteristics of the 104 enrolled patients at baseline are shown in Table 1. In total, 76% of the patients were men and 24% were black. The patients ranged in age from 28 to 83 years (median age, 57). A total of 87% of the patients had stage 5 chronic kidney disease, and 82% of the patients were undergoing hemodialysis at baseline. The mean estimated glomerular filtration rate at baseline among patients who were not undergoing hemodialysis was 20.6 ml per minute per 1.73 m2. A total of 19% of the patients had compensated cirrhosis at baseline, and 42% had received previous treatment for HCV infection; a majority of the patients had received a combination of interferon and ribavirin. Most of the patients (86%) had hypertension at baseline, 45% had preexisting anemia, and 41% had preexisting diabetes mellitus. Additional relevant baseline conditions that were reported in at least 10% of the patients are listed in Table S1 in the Supplementary Appendix. A majority of patients had HCV genotype 1 infection (54 of 104 patients; 52%); 17 patients (16%) had genotype 2 infection, 11 (11%) had genotype 3 infection, and 22 (21%) had genotype 4, 5, or 6 infection. Baseline resistance-associated polymorphisms in NS3 or NS5A were detected in 29% of the patients (28 of 96) for whom sequencing data were available.
Efficacy
Treatment with glecaprevir and pibrentasvir for 12 weeks resulted in a sustained virologic response in 98% of the patients (102 of 104 patients; 95% confidence interval [CI], 95 to 100) at 12 weeks (Table 2). No patients were reported to have virologic failure, although 2 patients did not have a sustained virologic response at 12 weeks for other reasons (additional safety information regarding these patients is described below). The rate of sustained virologic response at 24 weeks was 96% (100 of 104 patients; 95% CI, 95 to 100); both patients who had a sustained virologic response at 12 weeks but not at 24 weeks were lost to follow-up between posttreatment weeks 12 and 24.
Safety
Adverse events that were reported in at least 10% of the patients were pruritus (21 of 104 patients; 20%), fatigue (15 of 104 patients; 14%), and nausea (12 of 104 patients; 12%) (Table 3). The rate of adverse events among patients who were undergoing hemodialysis at baseline was 72% (61 of 85 patients), and the rate among patients who were not undergoing hemodialysis was 68% (13 of 19 patients). Serious adverse events were reported in 24% of the patients (25 of 104); none of the serious adverse events were considered by the trial investigators to be drug-related. No patients had adverse events of liver decompensation. Of the 2 patients who did not have a sustained virologic response at 12 weeks, 1 patient who was undergoing hemodialysis at baseline and had both compensated cirrhosis and underlying hypertension died after posttreatment week 2 owing to cerebral hemorrhage, which was assessed by the investigator as being unrelated to the trial drug. This patient had HCV RNA infection that was undetectable at the time of the patient's last visit (posttreatment week 2). The other patient had a history of gastrointestinal tract telangiectasia and had discontinued the trial treatment prematurely because of a nonserious adverse event of diarrhea (which was considered to be possibly related to the trial drug) at treatment week 4. This patient had HCV RNA infection that was undetectable at the time of treatment discontinuation but became detectable at posttreatment week 5. Three additional patients discontinued the trial treatment prematurely because of adverse events (1 patient at treatment week 8 because of pruritus; 1 patient at treatment week 10 because of pulmonary edema, hypertensive cardiomyopathy, and congestive heart failure; and 1 patient at treatment week 12 because of hypertensive crisis); all 3 patients had a sustained virologic response at 12 weeks. Cardiovascular-related serious adverse events were reported in 6 patients (Table S2 in the Supplementary Appendix); all 6 patients had preexisting hypertension. Mean systolic and diastolic blood-pressure values tended to decrease over the course of the trial period (Table S3 in the Supplementary Appendix); the rates of clinically relevant abnormal blood-pressure values that were reported during the trial are summarized in Table S4 in the Supplementary Appendix.
Clinically relevant laboratory abnormalities were rare, and no abnormalities in alanine aminotransferase of grade 2 or higher (i.e., postbaseline alanine aminotransferase levels of more than 3 times the upper limit of the normal range) were reported (Table 3). Grade 3 hemoglobin abnormalities (i.e., postbaseline hemoglobin levels of <8 g per deciliter) were observed in 5 patients; however, all 5 patients had entered the trial with a hemoglobin level of less than 10 g per deciliter (grade 2), and none of these patients discontinued the trial drug prematurely. One patient who had compensated cirrhosis and had been undergoing hemodialysis intermittently at baseline had an isolated grade 3 elevation in the total bilirubin level, which was due predominantly to an elevated indirect bilirubin level on day 15 of treatment without either an associated adverse event or a concomitant elevation in the alanine aminotransferase level. By posttreatment day 29, this elevation in the total bilirubin level had resolved without treatment. In addition, during the period between the predose baseline visit and the last trial visit after 36 weeks, there was no significant change in the mean (±SD) estimated glomerular filtration rate in the 19 patients who were not undergoing hemodialysis at baseline (20.6±8.0 ml per minute per 1.73 m2 at baseline and 20.2±2.8 ml per minute per 1.73 m2 at the last trial visit, P=0.53). The mean changes over time in the glomerular filtration rate and in the alanine aminotransferase level in the overall trial population and among patients who were not undergoing hemodialysis at baseline are summarized in Tables S5 and S6 in the Supplementary Appendix.
Virologic Resistance Testing
Baseline polymorphisms in NS3 or NS5A were detected in 28 of the 96 patients (29%) for whom viral sequencing data were available; 24 patients had polymorphisms in NS5A only (Table 1). No patients had virologic failure, despite the presence of baseline polymorphisms.
Pharmacokinetic Analysis
In a subgroup of 6 patients who were undergoing hemodialysis at baseline and who participated in intensive pharmacokinetic sampling, arterial (predialyzer) and venous (postdialyzer) drug exposure levels in plasma were determined for glecaprevir and pibrentasvir (Table S7 in the Supplementary Appendix). The observed maximum plasma concentration and the area under the plasma concentration-time curve from the start of hemodialysis to the end of hemodialysis were similar for the predialyzer samples and the postdialyzer samples for both glecaprevir (≤3% differences) and pibrentasvir (≤6% differences).
Discussion
Currently, ribavirin-free treatment options for patients with end-stage renal disease and concomitant HCV genotype 1 infection are limited, and no approved interferon-free and ribavirin-free treatment options are available for patients infected with HCV genotype 2, 3, 5, or 6.17,18 As a result, large numbers of these patients remain untreated.26 In this trial involving patients who were infected with one of the six major HCV genotypes, had stage 4 or 5 chronic kidney disease, and could have been undergoing hemodialysis at baseline, the rate of sustained virologic response was 98% (102 of 104 patients) after 12 weeks of treatment with coformulated glecaprevir-pibrentasvir. No patients were reported to have had virologic failure, regardless of whether they had cirrhosis or had received previous treatment for HCV. The regimen had an acceptable safety profile in this population of patients with numerous coexisting conditions.
Three adverse events (pruritus, fatigue, and nausea) were each reported in more than 10% of the patients. The rate of adverse events among patients who were not undergoing hemodialysis at baseline was similar to that among the patients who were undergoing hemodialysis. Patients with renal impairment are considered to be at high risk for cardiovascular events,11,12 and a majority of the patients enrolled in our trial (86%) had preexisting hypertension. A total of 24% of the patients had at least one serious adverse event, and 6 patients had serious adverse events that were cardiovascular-related. High rates of serious adverse events are common in this patient population; other trials of direct-acting antiviral regimens involving patients with severe renal impairment have reported rates of serious adverse events between 15 and 27%.19,21,27
Few patients discontinued treatment during our trial, and 3 of the 4 patients who discontinued treatment because of adverse events had a sustained virologic response at 12 weeks. The single patient who discontinued treatment and did not have a sustained virologic response at 12 weeks received treatment for fewer than 4 weeks.
Glecaprevir-pibrentasvir may fulfill an important unmet need; the absence of ribavirin as part of the treatment regimen minimizes the risks of treatment discontinuation and of adverse events due to anemia, which represents a considerable benefit for patients with severe renal insufficiency, since they are at increased risk for life-threatening anemia and cardiac events.28 In addition, the rate of grade 3 hemoglobin abnormalities that were reported in this trial was less than 5%, probably owing to the absence of ribavirin as part of the treatment regimen. Furthermore, although this patient population was expected to have a gradual decline in kidney function over the course of the trial, the mean decrease from baseline in the estimated glomerular filtration rate among patients who were not undergoing hemodialysis at baseline was minimal (0.4 ml per minute per 1.73 m2 at week 36). The data from the 19 patients who were not undergoing hemodialysis at baseline suggest that glecaprevir-pibrentasvir did not negatively affect kidney function.
Many direct-acting antiviral regimens are not recommended for patients with advanced renal impairment; the safety of regimens inclusive of sofosbuvir is not established in patients with severe renal impairment. Sofosbuvir-containing regimens are not recommended in patients with severe renal impairment because the principal metabolite of sofosbuvir (GS-331007) is excreted predominantly through the kidney, which leads to drug exposures that are up to 20 times as high as the intended exposure29; such patients have shown progressive deterioration of renal function when treated with a sofosbuvir-based regimen.30 In contrast, neither glecaprevir nor pibrentasvir is excreted through the kidney, and minimal change in drug exposure is observed in patients with advanced renal disease.24,25 In this trial, glecaprevir and pibrentasvir were not removed from plasma by hemodialysis, a finding that is consistent with the previous results in patients who were undergoing hemodialysis but did not have HCV infection.25
A limitation of this trial was the absence of a placebo control group; the inclusion of a placebo group would have helped clarify the contribution of potential treatment-related adverse events relative to the considerable underlying adverse-event profile observed in HCV-infected patients with advanced renal disease. In addition, although patients who had received previous treatment with sofosbuvir-containing regimens were eligible to enroll in this trial, only two such patients were enrolled (both had a sustained virologic response at 12 weeks), possibly because sofosbuvir-containing regimens are not recommended in patients with severe renal insufficiency.
In conclusion, the EXPEDITION-4 trial enrolled patients who represent a population with an unmet medical need in the treatment of HCV infection: patients who are in the advanced stages of chronic kidney disease and have HCV genotype 1, 2, 3, 4, 5, or 6 infection, including patients with compensated cirrhosis with or without previous treatment for HCV. Treatment with ribavirin-free, coformulated glecaprevir and pibrentasvir resulted in a high rate of sustained virologic response; no patient had virologic failure, irrespective of HCV genotype, the presence or absence of cirrhosis, or other baseline factors.
| |
| |
| |
|
|
|