| |
Tenofovir alafenamide versus tenofovir disoproxil fumarate: is there a true difference in efficacy and safety?
|
| |
| |
Download the PDF here
http://viruseradication.com/journal-details/Tenofovir_alafenamide_versus_tenofovir_disoproxil_fumarate:_is_there_a_true_difference_in_efficacy_and_safety^/
Journal of Virus Eradication April 2018 - Andrew Hill1* , Sophie L Hughes2 , Dzintars Gotham2 , Anton L Pozniak3
1 Department of Pharmacology and Therapeutics, University of Liverpool, UK 2 Faculty of Medicine, Imperial College London, UK 3 Chelsea and Westminster Hospital NHS Foundation Trust, London, UK
Published April, 2018
ABSTRACT
Background
Higher plasma tenofovir concentrations are associated with higher risks of renal and bone adverse events. The pharmacokinetic boosters ritonavir (RTV) and cobicistat (COBI) significantly increase plasma area under the curve (AUC) concentrations of tenofovir disoproxil fumarate (TDF), by 25-37%. When combined with RTV or COBI, the dose of tenofovir alafenamide (TAF) is lowered from 25 mg to 10 mg daily, but the TDF dose is maintained at 300 mg daily.
Objective
To assess the differences in safety and efficacy between tenofovir alafenamide (TAF) and tenofovir disoproxil fumarate (TDF) in regimens with and without the pharmacokinetic boosters RTV and COBI.
Methods
A PubMed/Embase search inclusive of dates up to 17 July 2017 identified 11 randomised head-to-head trials (8111 patients) of TDF versus TAF. The Mantel-Haenszel method was used to calculate pooled risk differences and 95% confidence intervals using random-effects models. A pre-defined sub-group analysis compared TAF with TDF, either when boosted with RTV or COBI, or when unboosted.
Results
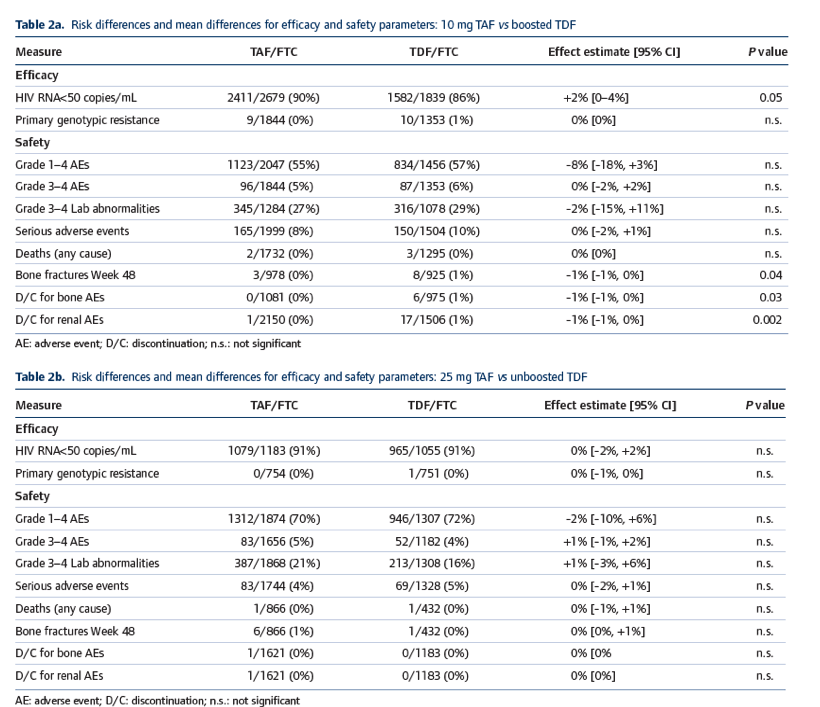
Nine clinical trials compared TAF and TDF for treatment of HIV-1 and two were for hepatitis B treatment. The eleven clinical trials documented 4574 patients with boosting RTV or COBI in both arms, covering 7198 patient-years of follow-up. Some 3537 patients received unboosted regimens, totalling 3595 patient-years of follow-up. Boosted TDF-treated patients showed borderline lower HIV RNA suppression <50 copies/mL (P=0.05), more bone fractures (P=0.04), larger decreases in bone mineral density (P<0.001), and more discontinuations for bone (P=0.03) or renal (P=0.002) adverse events. By contrast, there were no significant differences in HIV RNA suppression rates or clinical safety endpoints between unboosted TAF and unboosted TDF.
Conclusions
TDF boosted with RTV or COBI was associated with higher risks of bone and renal adverse events, and lower HIV RNA suppression rates, compared with TAF. By contrast, when ritonavir and cobicistat were not used, there were no efficacy differences between TAF and TDF, and marginal differences in safety. The health economic value of TAF versus low-cost generic TDF may be limited when these drugs are used without cobicistat or ritonavir.
Introduction
The nucleotide analogue tenofovir is used for the treatment of both HIV and hepatitis B, and for HIV pre-exposure prophylaxis. The original tenofovir disoproxil fumarate (TDF) version was developed at a dose of 300 mg once daily. A subsequent pro-drug formulation, tenofovir alafenamide (TAF), has recently been launched in North America and Europe at doses of 10 or 25 mg once daily. The pharmacokinetics of TAF lead to a 6.5-times higher intracellular concentration of the phosphorylated moiety tenofovir diphosphate, and 91% lower serum concentration of tenofovir, compared to TDF [ 1 , 2 , 3 ]. Given these pharmacokinetic differences, the dose of TAF can be far lower: a 25 mg once-daily dose of TAF is bioequivalent to TDF at 300-mg once daily, in terms of plasma levels of tenofovir. Pharmacodynamic studies suggest that the lower tenofovir concentrations in plasma produced by TAF translate to reduced off-target exposure to the drug in the kidneys and bones, for example, with implications for adverse effects [ 4 ]. TAF is therefore predicted to confer the same clinical efficacy as TDF, with potential improvements in tolerability [ 4 , 5 , 6 ].
The dose of TAF is adjusted from 25 mg to 10 mg daily in the presence of the pharmacokinetic enhancers ritonavir (RTV) or cobicistat (COBI) to account for their boosting effects [ 7 ]. Pharmacokinetic boosters also produce higher serum tenofovir levels when co-administered with TDF: the area under the curve (AUC) is 23% higher when TDF and COBI are co-administered, and 37% higher with atazanavir/RTV. However, adjustments to the 300-mg dosing of TDF are not made [ 1 , 8 , 9 , 10 ]. There could be other antiretrovirals causing changes in tenofovir exposure, such as rilpivirine, but doses of TDF again have not been adjusted to compensate.
TDF was first developed in clinical trials without the use of RTV or COBI. In these trials, the additional risks of bone or renal adverse events were small or non-significant, compared with other nucleoside analogues. For example, after long-term follow-up in the Gilead 903 study of the fixed-dose combination of TDF, lamivudine (3TC) and efavirenz (EFV), there were no discontinuations for bone or renal adverse events. There were small changes in bone mineral density in the first 48 weeks of treatment, but bone mineral density levels then remained stable for the next 2 years [ 11 ]. Similarly, there were no discontinuations for renal adverse events in several large clinical trials of unboosted tenofovir: Gilead 903 (TDF/3TC/EFV, n=299), Gilead 934 (TDF/FTC/EFV, n=257 [ 12 ]), ECHO/THRIVE (TDF/FTC/EFV or TDF/FTC/RPV, n=1096 [ 13 ]) and ASSERT (TDF/FTC/EFV, n=193 [ 14 ]). Also, long-term follow-up of patients treated with TDF or TDF/FTC for hepatitis B infection showed no significant change in bone mineral density for 3 years of follow-up [ 15 ].
A retrospective study of 7236 Veterans Health Administration patients found significantly higher risks of bone or renal adverse events for patients taking TDF with RTV- or COBI-containing regimens, versus TDF delivered unboosted, in combination with EFV. In this non-randomised observational study, patients using TDF with RTV or COBI had significantly higher risks of chronic kidney disease, proteinuria, osteoporosis and bone fractures [ 16 ]. In addition, analysis of pharmacokinetics has shown that patients with higher plasma concentrations of tenofovir are more likely to show renal impairment [ 17 ].
Most recently published randomised trials of TAF versus TDF have included RTV or COBI (e.g. with elvitegravir/COBI or darunavir/RTV). However, the most common use of TDF worldwide is unboosted, combined with either FTC or 3TC and either EFV or dolutegravir.
Previous meta-analyses of tenofovir safety have been conducted, but these have not divided studies by use of ritonavir or cobicistat [ 18 , 19]. The overall safety advantages of TAF may have been exaggerated by comparisons with a combination of boosted and unboosted TDF regimens [ 5 ]. This work aims to determine whether there are still safety advantages conferred by TAF when compared with unboosted as well as boosted TDF.
Methods
We conducted a meta-analysis of TAF versus TDF in treatment of HIV-1 and chronic hepatitis B in randomised head-to-head clinical trials. The databases PubMed, Embase and ClinicalTrials.gov were searched on 17 July 2017. The search terms for the intervention concepts were in accordance with the Cochrane guidance on search terms [ 5 , 20 ]. Studies qualifying for inclusion had randomised controlled designs, with at least 24 weeks of randomised treatment. Observational and dose-ranging studies were excluded [ 5 ].
Data were extracted on two measures of efficacy: patients with HIV RNA <50 copies/mL and treatment-emergent primary genotypic resistance. The efficacy results were analysed for the studies of either TAF or TDF in people living with HIV. The analyses of safety were conducted including studies of TAF versus TDF in either HIV or hepatitis B infection.
For safety measures, the risk of overall adverse events was assessed in terms of all-cause Grades 1-4 and Grade 3-4 adverse events, serious adverse events, Grade 3 or 4 laboratory abnormalities, and deaths (any cause).
Bone-related safety was assessed using the percentage change from baseline in hip bone mineral density (BMD), spine BMD, bone fracture events, and study discontinuations due to bone toxicity.
Renal outcomes included estimated glomerular filtration rate (eGFR) using the Cockcroft-Gault equation (mL/min), change from baseline in serum creatinine (mg/dL), percentage change in urine protein to creatinine ratio, urine albumin to creatinine ratio, urine retinol-binding protein (RBP) to creatinine ratio and urine beta-2-microglobulin to creatinine ratio, and study discontinuation due to renal toxicity (n=9). Studies with rilpivirine-containing regimens were excluded from analyses of creatinine parameters because of the independent effects of rilpivirine on creatinine levels [ 21 ].
Across studies, a mixture of means and medians were reported for many outcomes and were treated as equivalents. Confidence intervals and interquartile ranges, or statistical significance levels (P values) of the difference between groups were used to obtain standard deviations, applying methods recommended in the Cochrane Handbook [ 20 ]. RevMan Software Version 5.3 was used for all analyses.
The clinical diversity in TAF and TDF regimens and patient populations warranted the use of random-effects models, following Mantel-Haenszel methods [ 20 ]. Heterogeneity was assessed using the I2 statistic and was considered moderate from 30%≤I2<50%, substantial from 50%≤I2<75% and considerable when greater than 75% [ 20 ]. Pre-defined sub-analyses explored variation over follow-up time and differences by boosting of the TDF arm of studies.
Results
The literature search identified a total of 35 PubMed records, 148 Embase records and 70 clinical trials (Figure ()). Of these 253 records, 10 eligible randomised controlled trials comparing TDF with TAF had results. Results of a further eligible study were identified at the 9th International AIDS Conference on HIV Science. Reasons for exclusion include incorrect trial design [ 22 ] and short trial duration [ 23 ].
Trial participants were on average 83% male, 59% white, with a mean age of 41 years. Mean or median baseline CD4 cell counts at baseline were above 300 cells/μL in all of the trials. The 11 studies document 10,791 patient-years of follow-up, with 3347 patients receiving TDF and 4763 receiving TAF. A total of 4574 patients (7198 patient-years of follow-up) were allocated to boosted regimens, and 3537 patients (3594 patient-years) to unboosted regimens. 144-week data (the longest follow-up period) was available only for boosted studies.
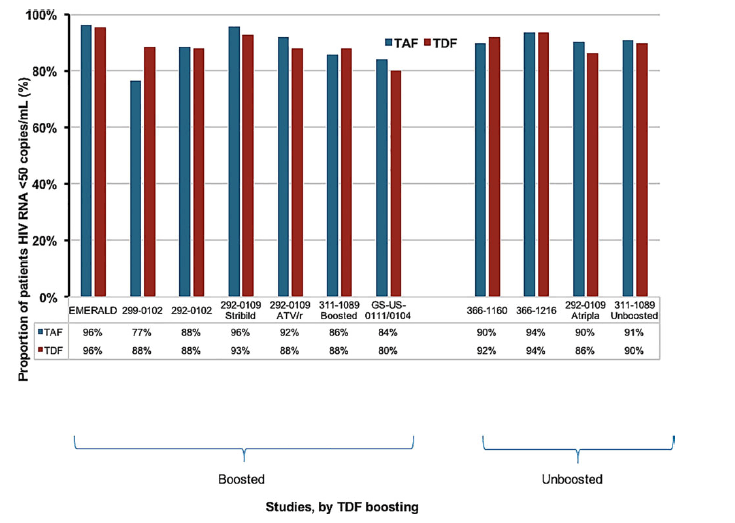
Efficacy outcomes
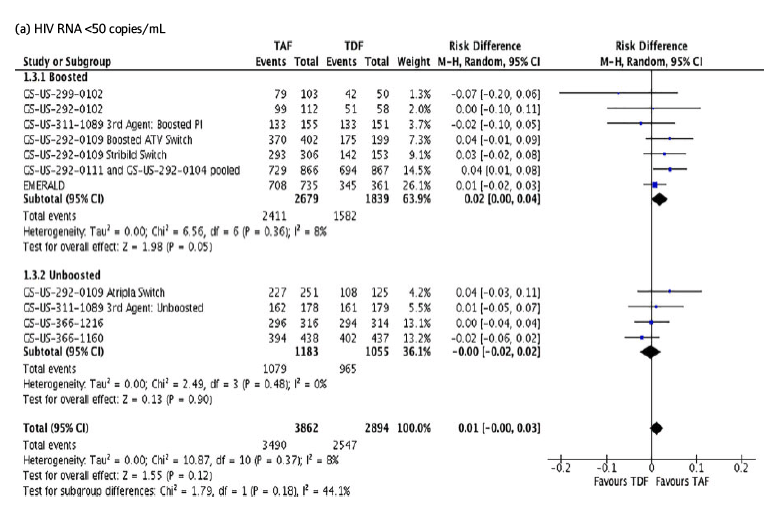
Figure () shows the percentage of patients with HIV RNA suppression <50 copies/mL, by treatment arm, at the end of each study. Overall, patients taking boosted TAF had 2% higher rates of HIV RNA suppression <50 copies/mL than boosted TDF (95% CI 0% to +4%, P=0.05). However, there was no difference in HIV RNA suppression rates <50 copies/mL between unboosted TAF and unboosted TDF (difference=0%; 95% CI -2% to +2%, P=0.90) (Figure ()a).
No significant difference in the emergence of primary genotypic resistance was detected between TAF and TDF using either random- or fixed-effects models (risk difference 0%, 95% CI 0% to 0%, P=0.80). No difference was observed between boosted and unboosted TDF regimens.
Figure 2. Summary of trial data for outcome HIV RNA <50 copies/microliter. Atripla: tenofovir disoproxil fumarate/emtricitabine/efavirenz; ATV/r: boosted atazanavir switch; Stribild: elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate
Clinical adverse events
As shown in Tables and , there were no significant differences between TAF and TDF for measures of Grade 1-4 or Grade 3-4 adverse events, serious adverse events, Grade 3-4 lab abnormalities or deaths, with no differences apparent between boosted and unboosted subgroups.
Renal parameters
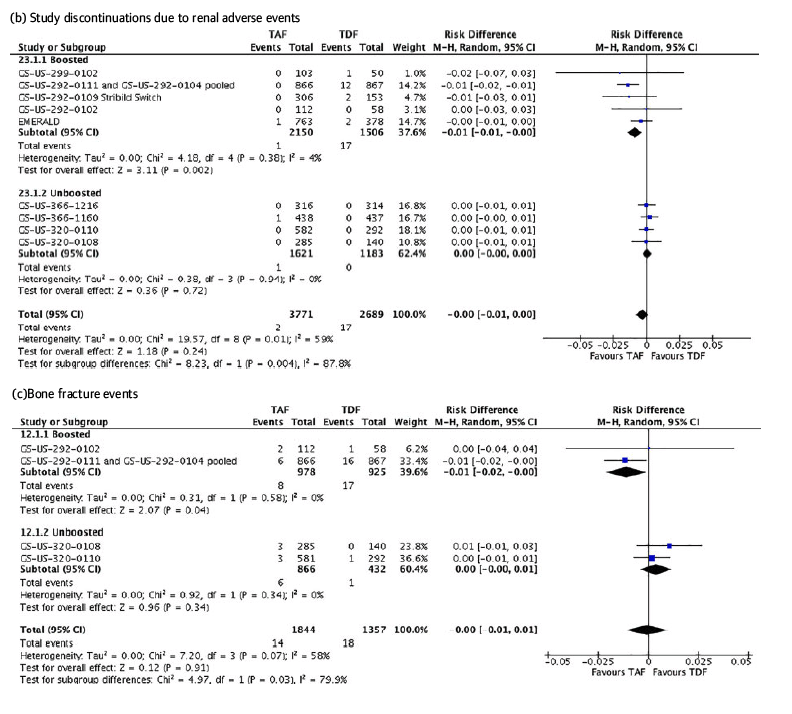
The test for differences by boosting revealed substantial heterogeneity between boosted and unboosted subgroups for study discontinuations due to renal toxicity through week 48 (I2=73%). The risk of discontinuation for renal adverse events was 1% lower for boosted TAF than boosted TDF (95% CI -1% to 0%, P=0.002). However, there was no difference in the risk of discontinuation for renal adverse events for unboosted TAF and unboosted TDF (difference=0%, 95% CI 0%).
Bone parameters
Across all follow-up times, the risk of bone fractures was 1% lower for boosted TAF compared with boosted TDF (95% CI -2% to 0%, P=0.04). However, there was no difference in the risk of bone fractures between unboosted TAF and unboosted TDF (risk difference=0%, 95% CI 0% to +1%) (Figure ()c). Similarly, patients taking boosted TAF were significantly less likely to discontinue treatment for bone-related adverse events than patients taking boosted TDF (risk difference -1%, 95% CI -1% to 0%, P=0.03). By contrast, there was no difference in the risk of discontinuation for bone-related adverse events between unboosted TAF and unboosted TDF (risk difference=0%, 95% CI 0%).
The test for differences by boosting revealed considerable heterogeneity between subgroups for percentage change in hip BMD at 48 weeks (I2=75.8%). In the subgroup of studies where TAF was compared with boosted TDF, percentage decreases in hip BMD were 1.98% smaller in TAF than in TDF (95% CI 1.63% to 2.34%, P<0.001), whereas in unboosted TDF regimens this mean difference reduced to 1.48% (95% CI 1.14% to 1.81%, P<0.001).
Differences were also observed between boosted and unboosted subgroups for spine BMD (I2=51.4%). TAF was associated with a 2.11% (95% CI 1.80% to 2.41%, P<0.001) smaller percentage decrease in spine BMD than boosted TDF whereas this mean difference reduced to 1.73% (95% CI 1.32% to 2.14%, P<0.001) when TAF was compared with unboosted TDF.
Figure 3. Forest plots of comparison through all follow-up periods for (a) Patients with HIV RNA <50 copies/mL through all follow-up periods, (b) study discontinuations due to renal adverse events, (c) bone fracture events.
Discussion
This meta-analysis of 11 trials, totalling 10,791 patient-years of follow-up, compared TAF with boosted and unboosted TDF. In randomised clinical trials where TAF and TDF were used without pharmacokinetic enhancers - ritonavir or cobicistat - there was no benefit of TAF versus TDF for HIV RNA suppression, clinical adverse events, discontinuation for renal adverse events, bone fractures or discontinuation for bone-related adverse events. By contrast, in randomised clinical trials where TAF and TDF were boosted by ritonavir or cobicistat, TAF showed significantly higher rates of HIV RNA suppression than TDF, and there were lower risks of renal and bone-related adverse events.
The meta-analysis adhered, where possible, to the methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions [ 20 ]. A highly sensitive search strategy was employed, covering PubMed, Embase and ClinicalTrials.gov. The selection criteria assured that only randomised clinical trials were included, limiting the possibility of bias. The study is of broad scope in terms of participants and interventions, which enhances the generalisability of the findings. Meta-analyses were conducted for all reported outcomes, providing a comprehensive summary of available evidence.
The clinical advantages of TAF over TDF depend on its more favourable bone and renal safety profiles. This analysis, however, shows that the benefits conferred by TAF may be systematically exaggerated by comparisons with a mixture of boosted and unboosted TDF regimens. There were still differences in bone mineral density between unboosted TAF and unboosted TDF, but the clinical significance of these differences is unclear. For context, the 48-week percentage change from baseline in lumbar spine BMD in HIV-positive youth is 1.15% when vitamin D3 is administered at a high dose: this is comparable with the difference between unboosted TAF and unboosted TDF [ 24 ]. The differences in mean bone mineral density scores were not associated with any significant increase in the risk of bone fractures or discontinuations for bone-related adverse events. In other long-term studies, unboosted tenofovir leads to a short-term reduction in bone mineral density, but there is then no change on long-term follow-up, for up to 7 years. Similar profiles are seen for creatinine clearance, with no progressive declines over time, after the initial changes within 24 weeks of starting unboosted tenofovir [ 25 ]. The results are also consistent with a pre-planned meta-analysis of the Gilead 102 and 103 studies. These studies randomised 1408 patients for 3 years of treatment with TDF/FTC/EFV, TDF/FTC/ATV/r or TDF/FTC/ELV/c. There were no discontinuations for renal adverse events among the 352 patients treated with unboosted TDF, as part of TDF/FTC. However, among the 1056 patients taking boosted TDF (with either ATV/r or ELV/c) there were 21 discontinuations for renal adverse events. The clinical advantages of unboosted compared with boosted TDF were also shown in the Veteran Affairs cohort study, which reported a 17% lower risk of chronic kidney disease for unboosted versus boosted TDF, and a 29% lower risk of osteoporosis [ 16 ].
The results from this meta-analysis are also supported by a biological mechanism: when 300 mg TDF is co-administered with COBI, the tenofovir AUC increases by 1.23 (1.16-1.38), and by between 1.22 to 1.37 for ritonavir-boosted PIs [ 1 ]. This increased tenofovir exposure is suggested to impact the kidneys and bones [ 5 , 16 ]. The dose of TAF is adjusted from 25 mg to 10 mg when used with ritonavir or cobicistat, to compensate for the boosting effects. It is unclear why the dose of TDF was never adjusted downwards as well, as part of the clinical development plan from the originator company Gilead. It would be possible to use lower doses of TDF - either 200 mg or 150 mg - when combined with ritonavir or cobicistat in a co-formulated tablet. It should be possible to re-design the co-formulations of TDF/FTC with darunavir/cobicistat or elvitegravir/cobicistat to improve their safety profiles.
Worldwide, the vast majority of TDF used is unboosted, typically as part of the combination pill TDF/3TC/EFV, available for under $100 in low- and middle-income countries (LMICs) [ 26 ]. A new co-formulated pill with TDF/3TC/DTG is being introduced in LMICs, at a cost of $75 per person-year [ 27 ]. In March 2018, the co-formulation of TAF/FTC/DTG was also launched at the same initial price [ 28 ]. The randomised ADVANCE trial is comparing TDF/FTC/EFV, TDF/FTC/DTG and TAF/FTC/DTG for 1110 treatment-naïve patients in South Africa, with 48-week results expected in the summer of 2019 [ 26 ]. The results from this meta-analysis would suggest that the difference in clinical safety and efficacy between TAF and TDF in this randomised study will be small, because both drugs will be given without either ritonavir or cobicistat.
The suitability of TAF for a new first-line regimen in developing countries depends on programmatic considerations including the possibility of harmonisation of use across subpopulations [ 26 ]. Thus far, gaps in the TAF evidence base on safety and efficacy in pregnancy and TB-co-infection have prevented its admission into WHO guidelines and the WHO Essential Medicines List [ 29 ]. Results are expected from trials to clarify the effectiveness and safety of TAF in pregnancy in mid-2019 [ 26 ]. Currently, the data on TAF in pregnancy includes only 12 live infants, including two congenital abnormalities. A further 17 induced terminations were documented, which included two additional congenital abnormalities [ 30 ]. The first results of trials of TAF and rifampicin in healthy volunteers suggest that intracellular concentrations of tenofovir diphosphate are maintained above those seen in patients given standard dose TDF [ 30 , 31 ]. It will be important to have clinical outcome data in people living with HIV co-infected with TB and treated with TAF-containing combinations while receiving rifampicin-based treatment for TB, to validate these results from healthy volunteers.
Other considerations in the transition to TAF from TDF in developing countries include cost and availability of fixed-dose combinations [ 26 ]. In countries included in the voluntary licence on TAF, registration of TAF-containing fixed-dose combinations should be prioritised. Boosting drugs are unsuitable for mass-production, limiting their suitability in low-income countries [ 1 ]. The ADVANCE and VESTED studies will address the need for comparisons of TAF with unboosted TDF, while comparisons with boosted TDF are of limited relevance in these settings [ 31 ].
The differences between comparisons of TAF with boosted and unboosted TDF are also of interest when the cost implications for middle- and high-income countries are considered. The only known economic analysis comparing TAF with TDF used input parameters derived from studies administering boosted TDF, which may not have been appropriate. It found a maximum willingness to pay for TAF of $990 higher than TDF in the USA [ 32 ]. Economic analyses should instead consider TAF against unboosted TDF to inform national policies. In the UK, the cheapest TAF regimen (TAF/FTC/RPV) costs $8246 PPPY, while the TDF patent is set to expire from 2018 across Europe [ 33 , 34 ]. In the generic-inaccessible context, money may be saved by using generic TDF-based regimens costing $107 PPPY [ 35 ].
This analysis aimed to inform policy discussion surrounding TAF, which has the potential to reduce the overall cost of treatment and facilitate expansion of ART coverage. However, the purported safety benefits of TAF over TDF may be overstated.
|
|
| |
| |
|
|
|