 |
|
|
| |
|
ICAAC Summary Report
|
| |
| |
Written by David Margolis, MD, University of Texas, Southwestern Medical Center and Dallas VA Medical Center
TOPICS
- treatment interruptions
- monotherapy Kaletra pilot study
- double boosted PI regimen without NRTIs: indinavir 800 mg bid plus Kaletra (lopinavir/rtv)
- new CCR5 co-receptor inhibitor (UK-427,857)
- T-1249, new fusion inhibitor
- new NNRTIs
- failure of ABC/TDF/3TC
- ABC/3TC/EFV once a day
- comparison of ABC vs AZT plus 3TC and EFV
- NNRTI resistance is not always predicted genotype
- ddI studied in patients with NRTI and ddI experience
- two NRTI resistance pathway - it may matter
- an unexpected interaction between amprenavir and lopinavir thwarts triple PI salvage
- pharmacology (drug interactions)
Monotherapy or no therapy at all: is this the new direction of HIV therapy?
As the pendulum governing prescription of antiretroviral therapy swings farther away from “hit hard and hit early” towards “the treatment is worse than the disease,” clinicians around the world are testing ways to minimize exposure to antivirals. Trials examining drug interruption or cycled therapy, as well as using fewer drugs, were presented. Clinicians should consider these concepts carefully, without forgetting the evolving benefits of potent, simpler, and less toxic drugs, or the difficulty of achieving full functional immune reconstitution once severe immunodeficiency has been allowed to develop, and recent evidence that minor populations of resistant HIV can expand when therapy is interrupted.
BASTA: when enough is enough
Maggiolo (Abstr. H-448) reported the interim findings of a treatment interruption study called “BASTA.” It was unclear to this author whether this was an acroynm, but it was said that in Italy “basta” means “enough” (as in “basta, basta, no more pasta”). Patients durably suppressed to HIV-RNA < 50 copies/ml with stable CD4 > 800 cells/ul were randomized (2:1) to either interrupt or to continue their ongoing treatment. Therapy was to be restarted if CD4 count <400 cells. Of 114 patients enrolled, 76 stopped therapy while 38 continued it. At roughly 20 months, about 25% of patients assigned continuous therapy had stopped therapy, presumably due to fatigue or toxicity. Multivariate analysis found that only the CD4 nadir value predicted CD4 cell decline (P< 0.001). For those whose lowest CD4 count was < 200 the median time to restarting therapy was 6.9 months. For those with a T-cell nadir of 200-350 cells therapy was restarted in a median of 14.1 months, and 17.8 months for those with a nadir of 350-500 T cells. No patient with a nadir of > 500 T cells had to restart treatment. This adds to the findings of previous clinic cohort observational studies that suggest that if one does not need HAART when HAART was started, it is safe to stop. Additional biomarkers that could aid in the identification of patients at low or high risk of progression off therapy are needed.
A second Italian group took a similar approach in an observational, retrospective, multicentre study (Abstract: H-856) of a group of 140 patients whose CD4 count pre-interruption was >500 cells/µL, HAART for >12 months (median of 3.5 years), and a CD4 nadir >250 cells/µL. Criteria for restarting treatment were: a confirmed CD4 count <350 cells/µL and the patient willingness. At treatment discontinuation, median CD4 count was: 804 cells/µL, median VL was 1.70 log 10 copies/mL.
75 of 137 (56%) were still off therapy at the time of the presentation. 24.3% had experienced a CD4 decline to below 350 cells/µL, and 22.1% restarted treatment before CD4 dropped below 350 cells/µL. The median time to restarting HAART for any reason was 104 weeks (95% CI: 79-169). Median time was only 52 weeks, however, if CD4 at nadir was > 350 cells/µL. Independent predictors of therapy resumption were: CD4 nadir; months with undetectable VL; slope of CD4 pre-ART and most recent VL vs the level at interruption.
A cost of interruption
But lunch is not free. Barreiro and colleagues presented findings for a cohort that had de-intensified therapy (Abstract: H-857) in which therapy was later re-intensified. 187 HIV+ patients on HAART (69 on PIs and 116 on NNRTIs), with CD4 counts >350 cells/ul and HIV-RNA <50 copies/ml substituted DDI 400 mg qd plus hydroxuyrea 500 mg bid for their NRTI+NNRTI or NRTI+PI regimen. HAART therapy was restarted for viral rebound >10,000 copies/ml. Overall, 21% of patients who moved to ddI-HU showed viral rebounds >10,000 copies/ml (19 on PIs and 20 on NNRTIs).
All patients who then resumed a PI-based regimen suppressed viremia to <50 copies/ml. However, 55% (16 of 29) who restarted an NNRTI failed to attain HIV-RNA <50 copies/ml (p<0.001). Genotypic analysis was not able to be performed in all failures, but NNRTI resistance did not predominate. However this observation does add to the concern that if NNRTIs are discontinued in a suboptimal setting, their therapeutic potency may be lost. It is still unclear if strategies such as stopping NNRTIs 1-2 days before the rest of the regimen is stopped will avoid the induction of NNRTI resistance.
Back to monotherapy?
Gathe in Houston, with collaborators in Dallas, challenged the field by testing the safety and efficacy of Kaletra (LPV/r) alone in treatment-naive patients (Abstract: H-845). Gathe pointed out that monotherapy, if it could be shown to be effective, could avoid many of the high costs and synergistic toxicities of combination antiretroviral therapy. It should be noted that if multiple nucleosides can effectively control HIV replication in some patients, PI monotherapy with a sufficiently high barrier to resistance might control HIV as well.
Consenting patients enrolled in this open-label, unsponsored trial. There were no CD4 or VL criteria for entry, and 28 males and 2 females (Hispanic 20%, Brown 20%) who entered had a mean CD4 = 169.5 cells/mL and VL = 262,020 copies/mL. Lopinavir/ritonavir was dosed in a weight-based fashion, 3 capsules BID or 4 capsules BID if >70kgs. Patients intensified therapy at week 12 with saquinavir or tenofovir/3TC if desired.
At 24 weeks (or last observation) only one subject had not achieved < 400 copies HIV RNA/ml. This subject had baseline VL 500,000 and had declined to a nadir of 1510, but rebounded to 4270 at week 32 when saquinavir was added. At that time genotype showed only L63P, phenotype was wild type, and an adequate LPV trough was measured. However, 2 subjects were lost to follow-up, one was deported, 2 stopped therapy due to GI intolerance, 1 was nonadherent, and one had TDF/3TC added when active HBV was discovered. So by ITT, 9 of 30 failures/VL > 400 were seen by week 24 (70%). 21 of 22 patients on treatment at 24 week had achieved < 400 copies/ml (95%). No other significant AE's were reported and no TG elevations > Grade 2 were observed.
CrixiLop: two boosted protease inhibitors alone (Indinavir + Kaletra)
Of potential relevance to the concept of PI monotherapy, the CrixiLop Cohort Study (Abstr. H-853) challenged the therapeutic potency of boosted PIs alone in a salvage setting. Staszewski and colleagues studied 23 heavily pre-treated HIV-positive patients in the Frankfurt HIV Cohort who were experiencing treatment failure with NRTI resistance or intolerance. Salvage therapy consisted of indinavir (800 mg bid) plus LPV/r (400/100 mg bid) without additional RTIs or any other ART. All but one subject were male, with median age 40 years, prior exposure to 10.5 drugs and 3.5 PIs, viral load 5.2 log copies/ml, CD4 count 116 cells/ul.
At a median follow up of 28 weeks, 17 (61%) of patients remained on IDV/LPVr. 8 discontinued due to IDV intolerance and 3 due to virologic failure. Viral load had declined 2.4 logs (range 1.30-5.6) and 1.9 logs (range 1.5-5.2) at weeks 12 and 24 respectively. The remaining 6 patients had a no persistent response. Median CD4 count rose 70 cells at wk 24. IDV dose was decreased in 6 patients, and LPV/r doses in 3 due to intolerance. Clearly many questions about the baseline PI sensitivity of these patients and the durability of this response need to be answered, but the initial virologic response in patients exposed to multiple PIs is impressive.
Incremental progress with new and old antiviral drugs
Despite the proliferating frequency of meetings, it was encouraging to reports of steady advances in the development of new antivirals. Most exciting was the progress of multiple entry inhibitor drugs, creating optimism that we are moving towards combination therapy at the point of virus entry into the T cell.
CCR5 chemokine HIV co-receptor blocker: first report of anti-HIV effect in HIV-infected people
HIV requires a second receptor in addition to CD4 to enter cells. Drugs that effectively block this interaction are hoped to add a new and highly potent class of antivirals to our armamentarium. Of the two most-often used chemokine receptors, CCR5 is the receptor primarily used by most HIV strains dominant in HIV-infected people, both during the initial process of infection, and throughout the course of disease. This receptor is therefore a therapeutic target of great potential, particularly as some immunologically healthy humans are born with the functional absence of CCR5. Blockade of CCR5 by a drug might therefore be well tolerated.
A safety study of Pfizer’s CCR5 antagonist in HIV-negative volunteers (Abstr. H-874) showed that steady state drug levels were reached within 7 days of dosing. 100mg and 300mg BID was well tolerated over 28 days of dosing. There were no serious adverse events reported, and all treatment related adverse events were of mild or moderate intensity. No clinically significant increases in any laboratory safety tests including haematology, clinical chemistry or lipid profiles, and no clinically significant changes in 12-lead ECGs, blood pressure, or heart rate were observed.
Pozniak and colleagues then reported on the short-term antiviral activity of UK-427,857 (abstract H-443). 24 asymptomatic, untreated HIV positive patients with CD4 counts >250 cells/mm3 and plasma viral load >5000 copies/ml were treated with UK-427,857 for 10 days. Volunteers were given 25mg once a day, 100mg twice a day or placebo for 10 days and were followed for 30 days after drug was stopped.
Prior to dosing, patients were selected for this study that had predominant circulating viral populations that used the CCR5 receptor, rather than the other major co-receptor CXCR4 (seen most often in individuals with advanced AIDS). A concern is that blockade of CCR5-using viruses may lead to the selection and outgrowth of X4-using viruses, which might result in more rapid disease progression.
UK-427,857 was well-tolerated with no severe or serious adverse events. EKG abnormalities, seen with some prior chemokine blocker candidates, were not found. Although the Pfizer team hopes to develop a drug without food restrictions, plasma levels are much higher when the drug is taken in the fasted state than with a high-fat meal. A study evaluating the impact of food on viral load reduction is ongoing. In this study, drug levels were also much higher and more stable at the 100 mg bid dose, rather than the 25 mg qd dose.
However, the 100mg bid dose of UK-427,857 yielded an impressive mean decrease in viral load of 1.42 log10 in only 10 days, with a mean decrease of 0.42 log10 at the 25mg QD dose. It was discovered that one patient harbored a mixed population of X4 and R5 viruses. This subject responded poorly to UK-427,857 monotherapy, and although viral load did not increase, a semi-quantitative assay suggested that the proportion of circulating X4 virus increased roughly 10-fold.
An accompanying presentation (Abstr. H-875) showed UK-427,857 to be potent inhibitor of R5 HIV-1 replication in the lab, and predictably inactive against X4 or R5X4 strains. The drug’s antiviral activity was unchanged despite increases in virus innoculum added to laboratory viral cultures, suggesting that viral load should not have a direct effect on efficacy in patients.
Recent laboratory studies suggest that R5 receptors are a limiting factor for HIV entry, and that multiple R5’s are need for the entry of a single viral particle. In other words, one individual may have 100 R5 receptors and another person may have 90 R5 receptors, but HIV may only need 50 R5 receptors for entry. Donor-to-donor PBMC variations (variations among study subjects) in host co-receptor expression did not affect potency in the study discussed in abstract H-875, indicating that “427” should be active regardless of individual variations in CCR5 expression or function. Currently, it appears that Pfizer must define the optimal dose for UK-427,857, and whether high levels of drug that saturate available R5 receptors are necessary, or whether sub-saturating doses will be equally clinically effective. However, the initial potent antiviral effects seen in short-term monotherapy are very encouraging. An important new antiretroviral may not be too far off.
T-20 to T-1249: a sequence of fusion inhibitors?
Lalezari (Abstr. H-444) presented further follow-up on the activity and effect of the use of the second-generation fusion inhibitor T-1249 in subjects who had failed the first generation fusion inhibitor T-20. Data on 25 patients was initially presented at CROI ’03. 53 patients failing T-20 and other antivirals with a median baseline HIV RNA of 4.97 log10 copies/ml received at least one dose of T-1249. These subjects had been failing T-20 for a median time of 66 weeks (range 28-165). All (52 or 53) whose virus could be amplified demonstrated T-20 resistance mutations and/or decreased susceptibility. Nevertheless the median HIV RNA decline was 1.26 log (95% C.I. 1.40 to 1.09) after 10 days of therapy. No serious adverse events (AEs) related to T-1249 were seen, and site reactions and AEs were similar to T-20. The authors suggested that this potent short-term response to T-1249 implied that sequencing of fusion inhibitor drugs after the development of resistance might be a viable clinical strategy. Certainly more long-term data is needed. Alternatively, as T-1249 moves forward in development, the first use of this more potent agent may be preferable.
In a related presentation, Trottier (Abstr. H-835) reported the 48-week response to Enfuvirtide (T-20) in the TORO Trials. As presented at CROI 2003 this winter, heavily pre-treated patients (median 12 ARVs) were randomized to an optimized background (OB) regimen or to OB + enfuvirtide (ENF; 90 mg sc BID). The overall median viral load and CD4 count at baseline were 5.1 log copies/mL and 92 cells/ul, respectively. At 48 weeks, 30.4% of patients (201/661) on ENF+OB had VL <400 c/mL by ITT analysis. Most of these had responded by this criterion at 24 weeks, and maintained response, but a few (4.2% or 28 patients) dropped below 400 copies after 24 weeks and remained below 400 copies at 48 weeks. This findings suggested a continuing clinical benefit of T-20 use with OB in this difficult-to-treat population. The challenges of a year of T-20 in the setting of advanced disease appear surmountable, and as with other antivirals, a clinical benefit may be reaped despite the gradual evolution of drug resistance.
Blocking the viral “handshake” with the T cell
Colonno and colleagues at Bristol first presented BMS-378806 (“806” for short) at the Resistance meeting in 2002. BMS is discontinuing 806 but continuing ahead with alternate candidates in their research and development program of CD4 receptor inhibitors. The novel 806 small molecule is a specific inhibitor of HIV-1 attachment that blocks the interaction of the binding pocket of the viral envelope Gp120 with the CD4 receptor. The group presented further basic laboratory studies (Abstr. H-877) aimed at illuminating the precise mechanism of blockade. Their elegant work showed that 806 binding induces changes in the shape of gp120. Further, the shape-shifting forced by 806 may also affect gp120’s ability to bind the CCR5 co-receptor. Lab studies suggested a synergistic antiviral effect when 806 was combined with a CCR5 chemokine co-receptor inhibitor or gp41 fusion inhibitor. Although BMS is switching the focus of their program, the possibilities are exciting.
Developing NNRTIs
Hazen and co-workers from GSK added to the story of this new drug class (abstr. H-445), as first reported at CROI ’03. GW8248 is a benzophenone and a potent NNRTI. A prodrug form of GW8248 with increased bioavailability and solubility is also under development. Like other NNRTIs, GW8248 inhibits HIV in the low nanomolar range. It is of great interest as it is active against a wide variety of NNRTI-resistant strains, including Y181C and K103N. GW8248 exhibited additive activities with other NNRTIs (NVP, DLV and EFV), was additive or synergistic with the activity of nucleosides, and generally synergistic with the activities of PIs.
To pose a very challenging laboratory test to GW8248, Hazen and colleagues tried to evolve a GW8248-resistant virus in the laboratory, starting with a K103N NNRTI pan-resistant virus. After 8 serially passages of K103N-infected cells in the presence of sub-inhibitory concentrations of GW8248, GSK scientists were able to grow out a virus encoding new mutations at V106I, P236L and E138K that was 50-fold resistant to GW8248. This is a glass half-full, as while this experiment shows that resistance can develop, the fact that 8 passages and 3 new mutations are required is encouraging.
The activity of capravirine (CPV), a novel NNRTI, was evaluated in combination with nelfinavir and two NRTIs in a Phase 2 study in HIV-infected, NNRTI-experienced patients (Abstr. H-871). It is hoped that CPV will have expanded activity against NNRTI-resistant HIV. Due to animal toxicities, this study was closed prematurely, but 36 patients chose to continue on open-label CPV. Of these 16 remained virologically suppressed (VL < 400 copies/mL) after 28 to 34 months of therapy. While 80% of the patient isolates demonstrated high-level resistance (>10-fold) to one or more of the approved NNRTIs, 70% remained susceptible (<10-fold resistance) to CPV. Despite its slow development, it is hoped that we will have more promising news about CPV soon.
Still learning how to use old drugs
Two late-breaking studies of Abacavir/3TC backbones
1): Failure of ABC/TDF/3TC:
Worrisome results of the Glaxo SmithKline study ESS30009 resulted in a Dear Doctor letter warning against the use of abacavir, tenofovir, and 3TC as initial therapy in drug-naïve patients. Gallant presented details of an unplanned interim analysis of this study as a late-breaker at the Wednesday afternoon oral session (H-1722a). Data on 194 enrolled subjects was reported. Subjects were >18 years old, < 14 days of prior ART, VL> 5000 copies, with any CD4 count. Most subjects were male, CD4 median between 250-280 cells, VL 4.5-4.7 logs. Subjects received ABC/3TC and tenofovir or efavirenz.
Due to several cases of early virologic non-response an interim analysis of failures was performed. Failure for this analysis were defined as < 2 log decline by week 8, or > 1 log rebound from VL nadir, or confirmed VL>400 after confirmed suppression to < 50 copies. By week eight 5 of 92 (5.4%) subjects in the efavirenz arm failed, while 50 of 102 (49%) in the tenofovir arm failed. Viral RNA measurements at biweekly intervals showed that several subjects (at least 8 of the 50) had no decline of VL at week 2, and many had viral rebound between weeks 2 and 4.
Although relatively few subjects with data beyond 12 weeks were presented, the performance of the efavirenz arm appeared similar to prior studies of EFV with 2 NRTIs, and similar to the identical regimen within the ZODIAC trial (discussed below), with 19 of 20 subjects who had reached 16 weeks suppressed to < 50 copies. However less than 30% of subjects in the tenofovir arm had achieved <50 copies by weeks 12 or 16 (about 18 and 4 patients respectively, at those timepoints).
Of the 36 genotypes available from failing patients in the tenofovir arm at week 12, 23 had both K65R and M184V RT mutations, and 13 had M184V alone. M184V is, of course, selected by 3TC and abacavir, and K65R by abacavir and tenofovir. Other abacavir-related mutations, V118I and Y115F, were infrequently seen (3 instances each).
This regimen should therefore not be used in naïve patients, at least until further data is available. Thus far, no failures in patients that did reach <50 copies have been observed. Multiple explanations for this dreadful result have been proposed. PK data presented elsewhere at this meeting would suggest that inadequate intracellular drug levels when abacavir is given once a day (Abstr. A-1797) or a drug-drug interaction between ABC and TDF (Abstr. A-1615) does not account for the failure of abc/tdf/3tc, but an intracellular interaction between the active metabolite of ABC, carbovir, and intracellular tenofovir cannot be excluded. The fact that significant resistance to all the drugs in this regimen is afforded to the virus once it acquires K65R and M184V appears likely to play an important role. The high incidence of K65R, with few other ABC mutations, suggests that it may be rapidly acquired by pre-existing viral variants that encode M184V under the dual selection pressure of ABC and TDF.
Although the hypothesis that nucleoside backbones should be constructed with drugs that select for divergent resistance pathways, ie. non-TAM resistance pathways, not selecting ABC and TDF with TAM resistance pathways (eg. AZT or D4T) is one that might have been made by the GSK marketing department-- the scientific and clinical data that supports such a hypothesis may be too reasonable to dismiss. As with several presentations at ICAAC, this study also reminds us that novel antiretroviral combination strategies need to be validated in careful clinical studies before wide use in the field. What looks good on paper or in the pill box may not provide the best therapy.
2) ABC/3TC/EFV once a day is “ non-inferior” to twice a day:
Looking forward to an ABC/3TC combination pill as a component of once-a-day regimens, Gazzard present another GSK study, ZODIAC, as a late-breaker at the Wednesday afternoon oral session (H-1722b). 770 well-matched patients with mean age 36, 54% white, 81% male, median VL 4.9 log copies, 44% > 100,000 copies took QD 3TC and EFV and QD or BID ABC with matched BID or QD placebo.
All the data presented was indistinguishable between the two arms, with an equivalent proportion achieving < 50 copies at week 48 by as-treated or intent-to-treat analyses (66-68% and 86-87% respectively). Adverse events were similar to those previously observed for these drugs. Using the relatively stringent case reporting criteria, suspected HSR reactions were reported in 7 and 9% of the subjects in each arm. Despite this somewhat high rate of suspected HSRs reported in an EFV-containing regimen, only 23 of the 770 subjects withdrew due to all adverse events combined (not all of which were HSR).
Less than 10% of subjects in each arm had virologic failure, and resistance profiles on more than half of these could not be obtained due to low viral load at time of failure. Most of the mutations that could be detected at failure were M184I/V and K103N, as expected. A few L74V mutations were detected, and one subject had K65R. All cases with genotypes obtained retained virus sensitive to AZT, D4T, and TDF by phenotype.
Due to the statistical analysis pre-planned for this study, QD ABC/3TC/EFV is said to be “non-inferior” to BID administration. This means that the 95% confidence interval around the percentage of patients responding to QD therapy by strict intent-to-treat analysis (58-71%) cannot be said to be meaningfully below that of BID therapy (60-73%). In this particular case a post-hoc analysis would likely demonstrate statistical equivalence.
Stronger is not Better? Abacavir is non-inferior to zidovudine
DeJesus reported the findings of a large phase III, international, multi-center, randomized, double-blind, study that compared the virologic response to abacavir to zidovudine when given with 3TC and efavirenz (Abstr. H-446). HIV-1 infected, ART naïve adults with baseline median HIV-1 RNA 4.79 log10 copies/mL, and median CD4+ cell count was 264 cells/mm3 were followed for 48 weeks. 39% of patients had baseline vRNA >100,000 copies/mL. Of the 649 subjects who received therapy, 70% vs. 69% of subjects in the ABC vs. the ZDV groups, respectively, achieved vRNA < 50 copies/mL by week 48. Virologic failure was infrequent and comparable between the treatment groups. It was reported that subjects receiving ABC group had a significantly better CD4 cell increase at week 48 (ABC, +209 CD4+ cells/mm3; ZDV, +155 CD4+ cells/mm3, p=0.005), despite the fact that the virological responses were similar. It is unclear if this is a meaningful or reproducible result.
As might be expected, the safety profiles were comparable over 48 weeks of randomized treatment, and both regimens were generally well tolerated. ABC hypersensitivity led to discontinuations, as did characteristic GI and other toxicities of AZT.
Viral decay, EFV vs NVP: Another demonstration that the in vitro or monotherapy potency of a single drug does not predict the potency of a regimen was culled from the 2NN data (Abstr. H-848). First phase viral decay rates (days 0, 3, 7, 14) of NVP vs EFV plus d4T and 3TC were indistinguishable. Viral decay rates respectively were NVP 0.24 logs/day, EFV 0.23, and NVP+EFV 0.23.
Hypotheses were put forth to explain why ABC, clearly a more potent drug in monotherapy studies than AZT, and known to maintain activity in the presence of more mutations than AZT, did not outperform AZT. A simple explanation would be that ABC toxicities led to more discontinuations or interruptions of therapy. However, these findings recall Gilead 903 in which tenofovir did not outperform D4T, and a viral dynamics study reported by the Aaron Diamond group last winter in which despite the use of >3 potent agents initial viral decays were not more rapid than standard therapy. Clearly, virological outcome is driven by multiple factors, and we do not understand all of these. Nevertheless, ABC/3TC is clearly another option as a dual nucleoside backbone. Longer term follow-up may find differences.
How much punch is left? the activity of DDI in experienced patients
To define the antiviral efficacy of DDI in antiretroviral-experienced patients, Molina and colleagues (Abstr. H-447) performed a 4-week study called Jaguar. Patients on stable therapy with plasma HIV RNA between 1000-100,000/ml were randomized (2:1) to add DDI (Videx EC qd 400 mg or 250 mg according to weight) or placebo to their regimen for 4 weeks. 168 pts received either DDI (n= 110) or placebo (n=58). At baseline, median HIV RNAlog10 was 3.8, median CD4 count was 378 cells/mm3. Median HIV RNA decrease from baseline in DDI and placebo arm were respectively -0.5 and -0.02 log10 at week 2 (p< 0.0001); - 0.59 and +0.07 at week 4 (p<0.0001). About 70% of patients in the study had prior ddI experience
Viral load dropped significantly despite the presence of up to 4 NRTI mutations, or 3 thymidine analog RT mutations. Although the numbers were small (17 subjects) a non-significant trend towards DDI effect was seen when 5 NRTI mutations were present, but not when 4 TAMS were seen. However, DDI retains significant antiviral activity in treatment-experienced patients with moderate RT resistance. This is clinically important information to use when trying to construct a salvage regimen.
Vive la Resistance
Weird viruses: NNRTI resistance is not always predicted genotype
Petropoulos (Abstract H-451) reported the rare discovery of viral isolates with high-level resistance to HIV-1 NNRTIs when assayed by Virologics phenotype despite the absence of known NNRTI resistance mutations. In a dataset of 18,034 samples, 48 isolates were identified that exhibited >10-fold resistance to at least one NNRTI in the absence of well-characterized NNRTI mutations (98G, 101E, 103N/S, 106A/M, 225H, 230L, 236L, or any mutation at position 100 181, 188, 190 or 227). Of these 10 encoded K101P and 13 the combination of K103R and V179D. The remainder of isolates contained other changes that have not yet been associated with NNRTI resistance. While this phenomenon is rare, it is useful to remember how much we have still to learn about the resistance of HIV to our drugs, and an illustration of the use of the phenotype assay to identify resistance not reported in a genotype assay. The reverse could also rarely occur, as it is conceivable that rare isolates with clinical resistance may not demonstrate significant fold resistance in phenotype assays.
Is there a clinical consequence to the “pathway” of resistance?
Under the selective pressure of a given drug combination, a specific pattern of resistance mutations tends to emerge if therapy fails. It is not yet known if a given pattern or “pathway” predicts a durable or transient response to second-line therapy. Such knowledge might guide decisions about initial and subsequent therapy.
Van Houtte and colleagues examined prevalence and phenotypic resistance of RT mutation combinations found in over 31,400 clinical viral isolates from routine testing at Virco between Jan `01 and Feb `03 (Abstr. H-907). Two “pathways” to resistance against NRTIs with mutations 41L, 210W, and 215Y/F or 67N, 70R, and 219Q/E/N/R in HIV-1 reverse transcriptase have been suggested. The prevalence and phenotypic resistance of these combinations (termed NAMs or nucleoside analog mutations) and the “non-pathway” 44D/E, 118I and 184V/I mutations in clinical isolates was examined. Resistance profiles came from Virco's matched genotype/phenotype dataset.
38% of isolates have > 1 NAMs (mean 2.7). Nearly half of these were phenotypically resistant to AZT (>4-fold). Median fold change in sensitivity to AZT in isolates with > 3 NAMs ranges from 3 to 37 when the 3TC resistance mutation M184I/V was present, and from 6 to 52 when it was absent. The 41-210-215 “pathway” predominates in this data set (34% of the isolates) vs. 67-70-219 (15%) with some overlap. Two, 3, and 4-NAM sets in the 41-210-215 pathway were on average more resistant to all NRTIs than those in the 67-70-219 pathway. Whether this predicts a poorer response to second-line therapy remains to be seen.
Tips and tidbits from pharmacology (Sessions A160 and A179)
- No PK interaction was observed when FTC was administered with tenofovir or zidovudine.
- In 35 day pk study of tenofovir and kaletra, PK of Lopinavir alone and RTV alone were unaffected by tenofovir. Tenofovir levels increased 32%, there were no serious adverse events reported. (ed note: In Study GS-908 TDF Compassionate Access Study (n=296), the incidence of confirmed changes in serum creatinine to >2.0 mg/dl or serum phospohorous <1.5 mg/dl was <1%. Five patients (1.8%; 5/271) experienced serum creatinine changes leading to TDF discontinuation: one patient developed Fanconi’s Syndrome (also experienced during prior use of high dose adefovir).
- In contrast to the observation that tenofovir increases the oral bioavailability of the Purine analog NRTI DDI, no increase in plasma level of abacavir was observed when tenofovir was administered. However, intracellular levels of carbovir, the active metabolite of abacavir, were not studied (see H-1722a above)
- Indinavir requires acid for absorption. Patients should be cautioned not to use omeprazole with IDV unless IDV/RTV is used. This may be particularly important as proton pump inhibitors may become OTC medicines soon. However, randitidine or Maalox did not affect levels of the amprenavir pro-drug 908, the investigation produg of amprenavir.
- Atorvastatin has no clinically significant effect on 908. Coadministration of 908, alone or with ritonavir, significantly increases atorvastatin exposure. Atorvastatin doses < 20mg/day should be used with 908 or another statin that is less dependent on CYP3A4 metabolism should be considered.
- Both tenofovir and UK-427857, an investigational antagonist of the CCR5 receptor, do not affect the activity of oral contraceptives
- Coadministration of the lower dose of 250 mg ddI-EC with 400 mg Atazanavir and 300 mg Tenofovir with food results in adequate DDI exposure. However, atazanavir levels are significantly reduced when given with tenofovir (with or without DDI). The addition of ritonavir may be required to overcome this, thus presenting challenges to the construction of a simple QD regimen that includes Atazanavir.
- Saquinavir/ritonavir 2000/100mg once a day achieves levels close to 1600/100 twice a day, and might be studied for use in patients without PI resistance. However, trough levels at the end of dosing are unacceptably low when 2000/100 is given, particularly so if PI resistance is present.
- The intracellular half-life of the active metabolite of abacavir, carbovir, was reported to be 20.6 hours. This would be sufficient for once a day dosing of abacavir, and suggests that a mechanism other than insufficient intracellular drug concentrations must account for virological failure in recent studies using once a day abacavir/tenfovir/3TC.
- Lopinavir levels were reported from assays of 31 CSF-plasma pairs from 26 HIV-infected individuals taking lopinavir-containing antiretroviral regimens. LPV was detectable in the CSF at concentrations that exceed those needed to inhibit HIV replication.
An unexpected interaction between amprenavir and lopinavir thwarts triple PI salvage
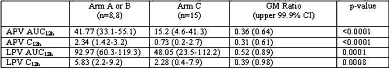
Several studies presented at ICAAC drove home the important lesson that we cannot do without careful testing of novel antiretroviral regimens. Drug interactions on many levels are complex and unpredictable, and the “one from column A and 2 from column B” approach cannot be depended upon. Adult ACTG Protocol A5143 was a carefully designed study to test the antiviral effect of combining GW433908 (Fosamprenavir or "908" the better absorbed prodrug of amprenavir nearing FDA approval) with Lopinavir/Ritonavir (LPV/R) in patient with PI resistance (Abstr. H-855a). PIs were given in combination with tenofovir plus 1 or 2 other nucleoside reverse transcriptase inhibitors (NRTIs).
Kashuba presented the findings of an open-label, steady-state PK substudy, performed in the absence of clear PK data to minimize subject risk. A planned independent interim review was performed after the first 8 subjects were randomized to each arm. Surprisingly, both APV and LPV exposures were substantially lower in the double PI arm (LPV 12 hr AUC 92.97 (60.3-119.3) vs. 48.05 (23.5-112.2) triple PI arm, and APV 12 hr AUC double PI 41.77 (33.1-55.1) vs. 15.2 (4.6-41.3) triple PI). Ritonavir exposure was similar in all arms and tenofovir did not account for the lowered PI exposure. This study was closed upon this analysis, and this combination of PIs should be avoided until further data is available.
Arm A (n=8): LPV/R 3caps BID Arm B (n=8): 908/R 700mg/100mg BID Arm C (n=17); LPV/R 3caps BID + 908 700mg BID.
|
|
| |
| |
 |
|
| |
|
|
|
|
|