| |
New targets and possible new therapeutic approaches in the chemotherapy of chronic hepatitis B
|
| |
| |
Hepatology, September 2003, Volume 38, Number 3
Jordan Feld1, Jia-yee Lee2, Stephen Locarnini2
1Department of Medicine, The Toronto Hospital, University of Toronto, Toronto, Ontario, Canada; and 2Victorian Infectious Diseases Reference Laboratory, North Melbourne, Victoria, Australia.
TOPICS
-Antiviral Therapy: Goals of Therapy
-The Viral Life Cycle: Where Are the New Targets?
-Pathogenesis of Ch-B: What Is the Host Doing?
-Possible New Targets, Therapies, and Directions
-Dynamics/Kinetic Studies and HBV: What Can be Learned?
-Conclusions: Is Combination Chemotherapy the Way Forward?
Chronic hepatitis B (CH-B) is characterized by inflammatory liver disease of variable severity driven by persistent replication of the hepatitis B virus (HBV). The development of a safe and effective hepatitis B surface antigen recombinant vaccine was an important milestone towards achieving control of CH-B, and its widespread implementation has dramatically reduced the incidence of infection. However, for those individuals chronically infected with HBV, antiviral chemotherapy represents the best prospect of controlling active replication and thereby preventing life-threatening hepatic disease.1 In this article, potential virus and host targets for possible future therapeutic intervention will be highlighted and reviewed, because existing therapies, either approved or in clinical trial, still have the disadvantage of low response rates and selection of resistance.
The complex interplay between the HBV-infected hepatocyte and the host immune response greatly influences the clinical course of disease and, consequently, strategies for clinical management. CH-B infection generally consists of an initial replicative phase with active liver disease developing during the elimination stage of hepatitis B e antigen (HBeAg)-positive CH-B, which involves HBeAg seroconversion and remission of liver disease. After HBeAg-seroconversion, a number of patients continue to experience active hepatitis due to the selection and persistence of HBV mutants that are unable to express HBeAg, a hallmark of HBeAg-negative CH-B.1 As morbidity and mortality in CH-B are linked to the development of cirrhosis and hepatocellular carcinoma, the goals of antiviral therapy are (1) to induce disease remission, (2) to arrest disease progression to cirrhosis, and (3) to block the sequelae of liver failure and/or hepatocellular carcinoma.
Goals of Antiviral Therapy
Interferon alfa (IFN-), lamivudine (LMV) and adefovir dipivoxil (ADV) are the only approved treatments for CH-B. While these treatments have similar short-term benefits in HBeAg-positive and HBeAg-negative CH-B patients, they have limited long-term efficacy. There is, therefore, a need for new treatment strategies for CH-B that can overcome some of these limitations. Several anti-HBV compounds are currently under evaluation (Table 1).
Table 1. Therapeutic Agents Available for Treating HBV Infections
|
| |
| |

|
| |
| |
Some of these including entecavir, emtricitabine (the 5-fluorinated derivative of LMV), clevudine (L-FMAU),3,4 and other L-nucleosides such as L-deoxythymidine (L-dT), are at various stages of clinical trials.
The efficacy of immunomodulatory approaches such as interleukin 2 (IL-2), IFN-y, thymosin or vaccine-based therapies have either been disappointing or have produced conflicting results. Therapeutic and DNA vaccine approaches, although theoretically attractive, have not progressed beyond clinical trials. Two pegylated interferons, PEG-IFN-2b (Peg-Intron) and PEG-IFN-2a (Pegasys), which have shown promise for the treatment of chronic hepatitis C,9 are currently being evaluated for CH-B. The future development of these approaches should provide clinicians with more therapeutic options.
While new therapeutic agents for CH-B are on the horizon, issues such as long-term efficacy and drug resistance will continue to pose challenges in patient management. Furthermore, it is not known whether combinations of antivirals and immunomodulatory agents will improve the clinical end points for CH-B.
Viral Life Cycle: where are the new targets?
The mature HBV virion is composed of a nucleocapsid core surrounded by a lipid bilayer incorporating the viral surface proteins. Residing within the nucleocapsid is the compact 3.2-kb partially double-stranded DNA genome that exists in a relaxed circular configuration with the genetic information organized into overlapping but frame-shifted open reading frames coding for the surface (L, M, S), core, precore (HBeAg), polymerase (pol), and the X proteins. An understanding of the HBV life cycle is crucial for the identification of potential antiviral targets.
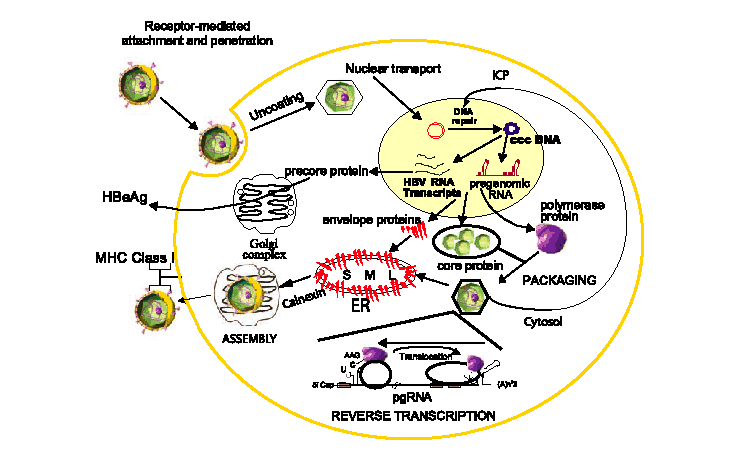
HBV replication begins when the virion attaches to an as yet identified receptor on the hepatocyte surface (Fig. 1). Following viral entry, the virus uncoats and is transported to the nucleus where the relaxed circular genome is converted by host cellular machinery to the covalently closed circular DNA (cccDNA); the cccDNA is, in turn, organized into viral minichromosomes. This key replicative intermediate is the transcriptional template for the production of the various HBV RNAs, including the pre-genomic RNA (pgRNA), that are necessary for viral replication and represents one of the major obstacles in the development of effective treatments for the control of CH-B.
Fig. 1. Diagrammatic representation of HBV replication in the context of its life cycle. The early events in the life cycle of receptor-mediated endocytosis and nuclear transport are not fully elucidated. The intracellular conversion pathway (ICP) where newly replicated HBV genomes are shuttled back to the nucleus for conversion to cccDNA to replenish the viral minichromosome pool is marked. The key virus-specific events of packaging and reverse transcription of the pregenomic RNA (pgRNA) are shown and expanded in Figs. 2 and 3, respectively. Modified from Delaney et al.
|
| |
| |
|
| |
| |
Following viral entry, the virus uncoats and is transported to the nucleus
where the relaxed circular genome is converted by host cellular machinery to the
covalently closed circular DNA (cccDNA); the cccDNA is, in turn, organized
into viral minichromosomes. This key replicative intermediate is the
transcriptional template for the production of the various HBV RNAs, including the
pregenomic RNA (pgRNA), that are necessary for viral replication and represents one
of the major obstacles in the development of effective treatments for the
control of
CH-B.
Pathogenesis of CH-B: What Is the Host Doing?
Because the liver disease caused by acute or chronic HBV infection is largely
immune mediated, understanding the complex and diverse immune responses to
HBV has potential immunotherapeutic implications. To this end, animal models
such as HBV-infected chimpanzees and HBV transgenic mice have led to significant
milestones in understanding the role of the immune system in causing liver
disease and in controlling and/or eliminating the virus. Although useful, there
are limitations to the use of animal models, particularly the HBV-transgenic
mouse model, which lacks the key HBV replication event, namely, the generation
and processing of cccDNA.
The course of HBV infection is influenced by the rigor of the immune
response. Activation of both the innate and adaptive immune responses during the early
stages of infection is critical to HBV clearance. Antigen-presenting cells
(APC), particularly dendritic cells, NK, and NKT cells, play an important role
in stimulating CD4+ helper T lymphocytes (Th) and CD8+ cytotoxic T lymphocytes
(CTL) for the control of HBV replication in the liver.
Th cells are divided into Th-type 1 (Th1) and Th-type 2 cells (Th2), based on
their cytokine-secreting profile. Th1 cells predominantly produce IL-2, which
induces the primarily cell-mediated immunity, whereas Th2 cells produce IL-4,
IL-5, and IL-10, which trigger the humoral immune response for the production
of antibodies. HBV clearance can be achieved when both the Th1 cells and CTL
are activated to generate a multispecific response to the viral envelope, pol,
and core proteins.
An immune response that predominantly activates Th2 cells for antibody
production most likely leads to HBV persistence. Thus, HBV persistence is
likely caused by a discrepancy in the T-cell repertoire resulting in tolerance to
HBV proteins during acute infection of neonates or adults. The simplistic
dogma of HBV clearance via cytokine-mediated destruction of HBV-infected cells
through CTL-activated perforin- or Fas-dependent pathways has now been
supplanted by a new paradigm placing emphasis on a cytokine-mediated noncytolytic HBV
clearance.
This new paradigm emerged through studies in acutely HBV-infected chimpanzees
15 and in HBV transgenic mice. Cytokines, predominantly tumor necrosis factor
a(TNF-a), IFN-g, and IFN a/b, were found to inhibit HBV replication without
necessarily killing infected hepatocytes. Induction of these cytokines in the
transgenic mouse model has been shown to interfere with at least two pathways
of the HBV life cycle: post-transcriptional degradation of HBV RNA 20 and
post-translational loss of HBV core protein via the elimination of immature
nucleocapsids containing pgRNA. Key cellular pro-cesses are likely to be involved.
IFN-appears to activate two cellular pathways involving dsRNA-dependent protein
kinase and dsRNA-dependent 2'5' oligoadeny-late synthetase. It is likely that
induction of PKR leads to inhibition of HBV protein synthesis, while
induction of 2'5' oligoadenylate synthetase activates RNase L to de-grade the HBV
RNA. Involvement of cellular processes was also shown in studies reporting IFN-a/b
Ð and IFN-gÐinduced changes to the transcription profile of cellular genes.
However, the molecular "antiviral" significance of these cytokine-induced
effects on HBV replication remains unclear.
The underlying molecular basis for the post-transcriptional degradation of
HBV RNA is emerging through HBV transgenic mice studies. These studies have
re-vealed a role for the cellular protein, La, in viral RNA degradation following
induction of TNF-a and IFN. In HBV, this protein binds specifically to a
predicted stem-loop structure located in the 5' end of the post-transcriptional
regulatory element within the viral RNA and appears to mediate the nuclear export
of the HBV RNA. Recent studies have suggested that specific HBV RNA
binding sites on the La protein could serve as potential antiviral targets.
The noncytolytic processes of the immune response for the control of HBV
replication in patients with CH-B are poorly understood. From a clinical mana
gement perspec-tive, the challenge will be to develop an immunotherapy strategy
that can overcome T-cell hyporesponsiveness with minimal infiltration of
inflammatory cells or liver cell necrosis, and to exploit noncytolytic clearance of
HBV from hepatocytes.
Possible New Targets, Therapies, and Directions
The review of the HBV life-cycle (Fig. 1) reveals that, apart from reverse transcription, most viral processes are dependent on host-cell machinery. The most important of these is the generation and persistence of cccDNA. Conventional antiviral inhibitors of viral DNA synthesis such as nucleoside/nucleotide analogues can prevent or reduce the development of new molecules of cccDNA. However, successful elimination of the existing pool of hepadnaviral cccDNA has only been achieved by either a noncytolytic Th1 immune response or immune-mediated cell killing followed by hepatocyte cell division. In this context, it is important to note that treatment of CH-B with nucleoside analogues can result in the (partial) restoration of (specific) immunoresponsiveness, which appears necessary for durable host-mediated control of infection. Collectively, the concept of successful therapy for CH-B is converging on the use of both antiviral and immunomodulating approaches.
Small Molecule Inhibitors
Polymerase Inhibitors: Nucleoside and Nucleotide Analogues. Nucleoside/nucleotide analogues are metabolized by cellular kinases into the triphosphate form, which can then selectively and competitively inhibit the catalytic properties of the viral polymerase. Incorporation of analogues that lack a functional equivalent of the 3’-hydroxyl group on the sugar moiety (such as LMV or ADV) results in chain termination of nascent viral genomes. Furthermore, purine nucleoside analogues can potentially inhibit the priming activity of the polymerase since the nucleotides present in the primed sequence of HBV minus-strand DNA are 5_-GAA-3_.
As shown in Table 1, a number of nucleoside/nucleotide analogues are being tested for use in CH-B. Except for entecavir, LY582563, and MIV-210, these compounds are in the unnatural “L” conformation. Thus, a major concern is that HBV-bearing LMV-resistance mutations such as rtL180M plus rtM204V/I may have significant cross-resistance to these L-nucleosides.
Viral Packaging Inhibitors. Several compounds have recently been developed that have a mechanism of HBV inhibition that is unrelated to the viral polymerase. The first of these are the phenylpropenamide derivatives, AT-61 and AT-130. King et al. showed that AT-61 did not affect total HBV RNA production or HBV DNA polymerase activity but did significantly reduce the pro-duction of encapsidated RNA. Importantly, both AT-61 and AT-130 have identical antiviral activity against wild-type as well as a number of different LMV-resistant strains of HBV. The mechanism of action of AT-130 has been examined by Feld and colleagues using the recombinant baculovirus system that packages HBVRNAin cis, and an HBV core protein expression vector system, which pack-ages in trans. In vitro studies using AT-130 showed significant inhibition of the production of encapsidated HBV RNA, but had no effect on total HBV RNA and did not affect core protein or nucleocapsid production, indi-cating an interference with the encapsidation process itself. Steric inhibition or interaction with the host cell chaperone proteins such as Hsp90, may be possible mechanisms.
Phenylpropenamides are not water-soluble and have extremely low bioavailability. Their future successful development as antiviral agents will depend on overcoming potential toxicity and medicinal chemistry issues. However, these observations represent an important proof of principle that both wild-type and drug-resistant HBV can be significantly and selectively inhibited at a level of replication that is independent of the viral poly-merase. A second class of compounds, the heteroaryldihydro-pyrimidines, are also potent non-nucleoside inhibitors of HBV replication both in vitro and in vivo. The heteroaryldihydropyrimidine compounds were developed by the Bayer Corporation and include the candidate molecule, Bay 41-4109, and congeners, Bay 38-7690 and Bay 39-5493. Exposure of HBV-infected cells to Bay 41-4109 resulted in increased degradation of core protein through improper formation of viral nucleocapsids. These HAP compounds were shown to have efficacy against HBV in the HBV-transgenic mouse model and to possess suitable preclinical pharmacokinetic and toxicology profile. Its novel mechanism of action and highly specific antiviral activity indicates that future clinical studies are warranted.
A third compound, LY582563, is a 2-amino-6-arylthio-9 phosphonomethoxyethylpurine bis (2,2,2-trifluoro-ethyl) ester, a novel nucleotide analogue derivative of phosphono-methoxyethyl purine, is in a structural class that is similar to ADV. This compound has excellent antiviral activity against HBV with a good preclinical toxicity profile. It is also effective against LMV-resistant HBV and its mechanism of action and early clinical development are under investigation by the Eli Lilly and Mitsubishi Pharma Corporation.
Gene Therapy Approaches: The Promise of Gene Silencing
Gene therapy is defined as the introduction of new genetic material into a target cell with a therapeutic benefit to the individual. Several genetic antiviral strategies including ribozymes, antisense oligonucleotides, interfering peptides or proteins, and therapeutic DNA vaccination have been explored for the molecular therapy of CH-B. Furthermore, molecular strategies to treat or prevent the long-term sequelae of CH-B–related cirrhosis and HCC are being investigated.
A novel molecular strategy that holds promise is the use of small interfering RNAs (siRNA). RNA interference is a cellular process of sequence-specific gene silencing in which small duplexes of RNA target a homologous sequence for cleavage by cellular ribonucleases. The introduction of approximately 22-nt siRNAs into mammalian cells can lead to specific silencing of cellular mRNAs with-outinduction of the nonspecific interferon responses that are activated by longer RNA duplexes. Post-transcriptional gene silencing mediates resistance to both endogenous, parasitic, and exogenous pathogenic nucleic acids and can regulate the expression of protein-coding genes.
This approach has been successfully applied to HBV and hepatitis C virus replicon RNAs in cell culture. The siRNA molecules dramatically reduced virus-specific protein expression and RNA synthesis, and these antiviral effects were independent of IFN.
While the approach of siRNA, along with conventional gene therapy approaches, holds great promise, issues such as gene delivery, stability, toxicity, resistance, and safety need to be resolved.
Immunomodulatory Approaches to Controlling HBV Infection
The goal of immunomodulatory therapy is to stimulate the host’s immune response to HBV-infected cells, and thereby terminate/eliminate viral infection. As IFN-a and thymosin-a have been reviewed elsewhere, this review will highlight other potential immunomodulatory agents for the treatment of CH-B.
IL-12 and IL-18. IL-12 and IL-18 are secreted by activated macrophages and dendritic cells. IL-12 is a cytokine that plays a critical role in the regulation of the immune system by promoting a Th1 response. IL-18 is an IFN-g–inducing factor that can act on Th1 cells, NK cells, and dendritic cells to produce IFN-g in the presence of IL-12. The significance of the Th1 versus Th2 response with respect to HBV infection has been discussed in the section on pathogenesis of chronic hepatitis B. Experiments conducted in HBV-transgenic mice have indicated that IL-12 injection results in a marked down-regulation of viral replication in the liver and extrahepatic sites. In this model, IL-12 appears to exert antiviral effects indirectly by inducing lymphocytes locally to secrete IFN-g, IFN-a/b, and TNF-. Examination of patients undergoing IFN-a therapy has revealed that levels of IL-12, as well as IL-2 and IFN-a (also Th1 cytokines), were significantly higher in patients that responded to IFN-a therapy in comparison to nonresponders. A recent phase I/II study of IL-12 in CH-B patients showed that doses of 0.25 and 0.5 ug/kg produced a significant reduc-tion in serum HBV DNA after 12 weeks of treatment.
However, the overall antiviral efficacy was lower than that reported for IFN-a or LMV. Importantly, this study showed that a short course of IL-12 therapy was safe, and with similar side effects to those observed with IFN-a therapy.
In transgenic mice studies, Kimura et al. showed that IL-18 inhibits HBV replication in the liver, via a noncy-topathic process that is mediated by IFN-g and IFN-a/b. These investigators also showed that IL-18 can synergize with IL-12 to inhibit HBV replication, indicating that coadministration of IL-18 and IL-12 may have therapeutic potential for the treatment of CH-B.
Dendritic Cell Vaccination. Dendritic cells play a vital role as APCs in stimulating adaptive immunity during viral infection as they prime naïve CD4+ and CD8+ T cells to produce cytokines that affect the Th1/Th2 immune balance. To serve as APCs, dendritic cells internalize endogenous or exogenous antigens, which are processed in the MHC class I or II pathways for presentation to T cells. In addition to their APC function, activated mature dendritic cells secrete a variety of Th1 and Th2 type cytokines including IFN-a/b,TNF-a, IL-1, IL-8, and IL-12.
Information on the role of dendritic cells in influencing Th1 or Th2 response during HBV infection is limited. It is likely that impairment of dendritic cell function may contribute to HBV chronicity.46,47 Interestingly, while dendritic cells act as potent stimulators of immunity, these cell can also induce immune tolerance through T-cell anergy or depletion.48 Dendritic cell–mediated tolerance is not well understood, and it remains to be determined whether these cells contribute to neonatal tolerance leading to HBV persistence.
The effectiveness of dendritic cells in manipulating the host immune response is currently being exploited clinically. The ex vivo culture and priming of human dendritic cells with antigens such as recombinant tumor and viral antigens, followed by autologous transfusion has been explored as a form of immunotherapy in several clinical trials. The role of dendritic cell immunotherapy for CH-B is still in its infancy, and strategies for the priming of dendritic cells with appropriate HBV proteins that will overcome immunologic tolerance and induce a multi-specific immune response to HBV will need to be investigated.
Dynamics/Kinetic Studies and HBV: What Can Be Learned?
Studies of viral dynamics in acute or chronic HBV have been performed and have provided important in-sights into the pathogenesis of host-virus responses. Furthermore, improvements in HBV DNA testing technology and mathematical modelling have resulted in several important advances in the field.
The pathogenesis of CH-B is characterized by a dynamic equilibrium between viral production and clearance. The introduction of antiviral therapy can upset this equilibrium by inhibiting virus production and causing a decline in the viral load. Indeed, the rate of decline in the viral load is a measure of the rate of viral clearance and by inference, must be equivalent to the rate of virus pro-duction before therapy. The various models proposed to describe the fall in serum HBV differ principally in their underlying assumptions regarding at least three factors: the nature and efficacy of the inhibition of viral replication that is imposed on the virus-host system by nucleoside analogue therapy, the behavior of the infected cell population after the commencement of therapy, and ultimately the residual effect of this population on the level of viremia.
Tsiang et al. followed the decline in serum virus caused by ADV monotherapy in patients with CH-B, and quantified the level of serum HBV using the Roche MONITOR PCR-based assay (Roche Diagnostics, Branchburg, NJ). These investigators reported a consistent biphasic decay in treated patients. Regression analysis allowed the calculation of a mean viral half-life of 1.1 _ 0.3 days, a mean infected cell half-life of 18 +/- 7 days, and a mean antiviral efficacy of 0.993 +/- 0.008. Lau et al., using the Digene Hybrid Capture II assay (Digene Diagnostics, Beltsville, MD) to monitor viral load, com-pared the effect of LMV monotherapy and LMV plus famciclovir (FCV) combination therapy on the treatment dynamic equilibrium. A mean virus half-life of 2 days and a mean infected cell half-life of approximately 40 days were calculated. The antiviral efficacy of the combination therapy was found to be significantly greater than that of LMV monotherapy.
More recently, Lewin et al. showed that the viremia of CH-B during potent combination antiviral therapy could not be entirely described by biphasic curves. Once therapy was initiated, there was a mean delay of 1.6 (from 1 to 5) days, before an antiviral effect could be detected, and in some patients, an increase in viral load was seen, after which a biphasic or typically a multiphasic decay of viremia occurred.
In most patients, two patterns for the first phase elimination (decay of viremia) was observed with either a rapid (t of 1.0 hour) or a slow (t of 92 hours) slope being observed. This was not related to pretreatment viral load, serum alanine amino-transferase level or the type of antiviral therapy (LMV monotherapy vs. LMV plus FCV combination therapy).
For these patients, the second phase elimination (elimination of infected cells) was then either flat or slow with a T= 7.2 +/- 1.2 days. In some patients, a complex or “stair-case pattern” was seen with further phases of viral decline or phases with little change in viral load. These complex decay profiles could possibly represent impaired cytolytic and noncytolytic mechanisms of in-fected cell loss and highlight the complexity of the CH-B carrier, reflected in the outcome of the virus-cell-host immune response interaction. However, these profiles do need to be better understood in terms of developing alter-native therapeutic approaches to improve the management of HBV-infected individuals.
Conclusions: Is Combination Chemotherapy the Way Forward?
The emergence of drug-resistant HBV as a consequence of long-term antiviral therapy and the low sustained/ durable responses of existing therapies signal the need for new approaches. Viral dynamic studies have pointed to a problem with the second phase of HBV decline that follows on from the initial rapid response to chemotherapy. This will be a major challenge to over-come, but new insights into HBV pathogenesis and the development of novel immunotherapies may provide useful ways forward. Whether this is by using additional nucleoside/nucleotide analogues or adding existing immunemodulators to nucleoside/nucleotide analogue combinations is, at this time, unknown but warrants further clinical investigation. The next logical step may be to initiate trials of triple therapy with two nucleoside/nucleotide analogues in combination with PEG-IFN. In the meantime, the development of the future generation of anti-HBV antiviral agents including non-nucleoside ana-logues such as packaging inhibitors and immunotherapies such as dendritic cell vaccines, may well be needed to finally achieve “a cure” for CH-B.
|
|
| |
| |
|
|
|