 |
|
|
| |
Efavirenz (Sustiva)-Based HAART: 168 Weeks of Follow-Up (3-4 yrs) of original 006 EFV pivotal Study
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
Reported by Jules Levin
*K Tashima1, S Staszewski2, M Nelson3, A Rachlis4, D Skiest5, R Stryker6, L. Bessen7, V Wirtz7, S Overfield7, D Sahner7
1Miriam Hospital, Providence RI, 2Goethe University, Frankfurt Germany, 3Chelsea and Westminster Hospital, London England,
4Sunnybrook & Women's College Health Sciences Centre, Toronto Canada, 5The University of Texas Southwestern Medical Center, Dallas TX,
6Tower I.D. Medical Associates, Los Angeles CA, 7Bristol-Myers Squibb Pharmaceutical Research Institute, CT and NJ USA
SUMMARY:
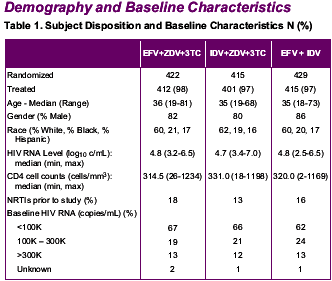
This is a 3/4 year update on the original 006 efavirenz study.
--Over 1200 patients randomized to EFV+AZT/3TC or IDV+AZT/3TC or EFV/IDV.
--KEY BASELINE CHARACTERISTICS: age: 35 yrs; 80% men; 60% white; 20% black; 16% Hispanic; HIV RNA: 4.7 log copies/ml; CD4 count: 320 cells; NRTIs prior to study: 13% in IDV arm, 18% in EFV arm; stratified baseline HIV RNA: <100,000 c/ml, 66%; 100,000-300,000: 20%; >300,000: 12%.
EFFICACY RESULTS:
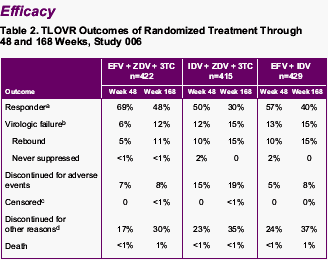
Time to loss of virologic response (<400 c/ml):
RESPONDERS-
--EFV+AZT/3TC: 69% at week 48, 48% at week 168
--IDV+AZT/3TC: 50% at week 48, 30% at week 168
--EFV/IDV: 57% at week 48, 40% at week 168
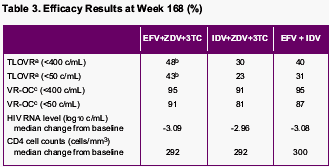
<50 copies/ml at Week 168:
TLOVR:
--EFV+AZT/3TC: 43%
--IDV+AZT/3TC: 23%
--EFV/IDV: 31%
VIROLOGIC FAILURE-
--EFV+AZT/3TC: 6% at week 48, 12% at wk 168
--IDV+AZT/3TC: 12% at wk 48, 15% at wk 168
--IDV/EFV: 13% at wk 48, 15% at wk 168
VIRAL REBOUNDS-
--EFV+AZT/3TC: 5% at wk 48, 11% at wk 168
--IDV+AZT/3TC: 10% at wk 48, 15% at wk 168
--IDV/EFV: 10% at wk 48, 15% at wk 168
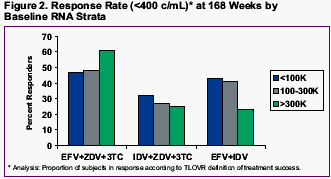
RESPONSE RATES (<400 c/ml) at 168 Wks by BASELINE HIV RNA-
<100,000 copies/ml-
--EFV+AZT/3TC: 47%
--IDV+AZT/3TC: 31%
--EFV/IDV: 45%
100,000 to 300,000 copies/ml-
--EFV+AZT/3TC: 47%
--IDV+AZT/3TC: 28%
--EFV/IDV: 42%
>300,000 copiesml-
--EFV+AZT/3TC: 61%
--IDV+AZT/3TC: 25%
--EFV/IDV: 23%
DISCONTINUED for ADVERSE EVENTS-
--EFV+AZT/3TC: 7% at wk 48, 8% at wk 168
--IDV+AZT/3TC: 15% at wk 48, 19% at wk 168
--IDV/EFV: 5% at wk 48, 8% at wk 168
DISCONTINUED FOR OTHER REASONS-
--EFV+AZT/3TC: 17% at wk 48, 30% at wk 168
--IDV+AZT/3TC: 23% at wk 48, 35% at wk 168
--EFV/IDV: 24% at wk 48, 37% at wk 168
INTRODUCTION & STUDY OBJECTIVES
The main goal of antiretroviral therapy is to obtain maximal suppression of viral replication for as long as possible. In order to accomplish this, antiretroviral regimens must be acceptably tolerated, durably potent, and simple enough to permit adequate adherence. Efavirenz is a proven component of standard of care in first line therapy and is a preferred ARV in HIV treatment guidelines. AI266006 was an open-label, randomized study designed to compare the antiretroviral activity and tolerability of three antiretroviral regimens, EFV + ZDV + 3TC, IDV + ZDV + 3TC, and EFV + IDV, in HIV-infected, NNRTI-, 3TC-, and PI-naïve subjects. The original study was amended to increase sample size and duration of follow-up, thus enabling assessment of the long-term durability of viral load suppression among treatment groups. The major objectives of the final analysis were to assess long-term efficacy and safety of the three treatment regimens. EFV-based therapy was compared with an IDV-containing triple therapy regimen considered to be standard of care at the inception of the trial.
METHODS
Subjects enrolled in the study were to be HIV-infected with measurable viral loads above 10,000 c/mL and CD4 cell counts >=50 cells/mm3. Follow-up was to continue until the last subject enrolled had completed 168 weeks on-study. The primary longterm efficacy analysis evaluated the proportion of subjects in virologic response (<400 HIV RNA c/mL) at 168 weeks according to the TLOVR (time to loss of virologic response) definition of treatment success; this ITT analysis defines response as a minimum of two sequential HIV RNA measurements < Level of Quantification (LOQ) maintained through the end of study without intervening replicated rebound, death, loss to follow-up, or discontinuation of protocol-assigned initial study therapy. Subjects who elected not to continue in the long-term extensions of the study were censored at the date of their last on-treatment viral load measurement. Secondary objectives assessed long-term HIV RNA suppression to levels below the LOQ (50 c/mL) of an ultrasensitive PCR assay, safety and tolerability.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
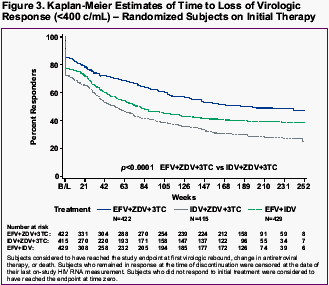
a -Patients achieved and maintained confirmed HIV RNA <400 copies/mL through Week 48 or Week 168.
b -Includes confirmed viral rebound and failure to achieve confirmed HIV RNA <400 copies/mL through Week 48 or Week 168.
c -Patients with HIV RNA levels <400 copies/mL who chose not to continue in the voluntary extension phases of the study were censored at date of last on-treatment viral load measurement.
d -Includes consent withdrawn, lost to follow-up, noncompliance, never treated, missing data, protocol violation, lack of effect/therapeutic failure, and other reasons.
|
|
| |
| |
 |
|
| |
| |
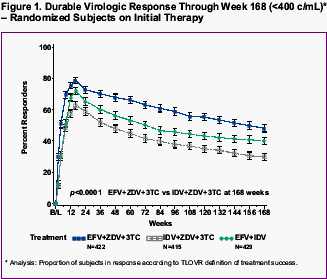
a -Proportion responding using the TLOVR definition of response
b -p<0.0001 for difference estimate (versus response in IDV/ZDV/3TC control group)
c -Virologic response (observed cases)
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
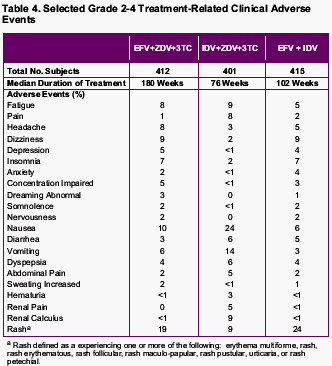
Tolerability and Safety
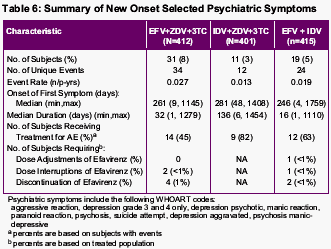
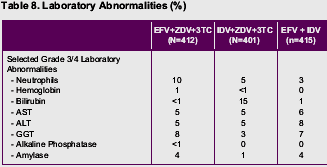
No unexpected long-term safety concerns emerged in any treatment group. New-onset adverse events that occurred with greater frequency in subjects receiving efavirenz typically occurred relatively early after the initiation of therapy.
Treatment-related grade 3/4 adverse events were more frequent in subjects receiving indinavir-containing therapy than in those receiving efavirenz-containing regimens (34% vs. 22%, efavirenz plus indinavir; 24%, efavirenz-containing triple therapy).
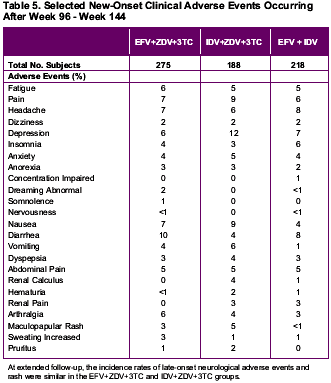
New-onset adverse events were evaluated by time period on-study (0 to 24 weeks, more than 24 to 48 weeks, more than 48 to 96 weeks, more than 96 to 144 weeks, and more than 144 weeks). During the first 24 weeks of the study, new-onset adverse events occurring with greater frequency (>=5% difference) in either of the efavirenz treatment groups relative to the control group were dizziness, insomnia, impaired concentration, abnormal dreams, nervousness, and maculopapular rash. New-onset adverse events occurring with greater frequency (>=5% difference) in the indinavir-containing triple therapy group were nausea, vomiting, hyperbilirubinemia, and pain.
After 24 weeks, subsequent time periods showed no new-onset adverse events occurring with greater frequency (>=5% difference) among efavirenz-treated subjects compared with the indinavir triple therapy group. In contrast, nausea, hyperbilirubinemia, upper respiratory infection, dry skin, renal calculus, coughing, depression, pain, and arthralgia emerged more frequently in subjects receiving indinavir-containing triple-drug therapy during one or more subsequent time periods.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
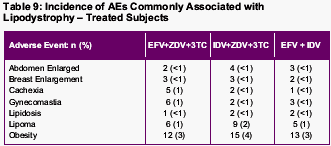
Lipodystrophy
Lipodystrophy was not systematically assessed in subjects by objective measures. Lipodystrophy (of any severity, regardless of relationship to study therapy) was reported as an AE in 4%, 3%, and 5% of subjects treated with EFV + ZDV + 3TC, IDV + ZDV + 3TC, and EFV + IDV, respectively. The frequencies of other AE terms that may be associated with lipodystrophy are presented below.
|
|
| |
| |
 |
|
| |
| |
|
| |
|
|
|
|
|