 |
|
|
| |
CROI Report: Pharmacology of HIV Drugs
|
| |
| |
Written for NATAP by Peter Anderson, PhD
University of Colorado
The 11th CROI had approximately 40 posters and oral presentations, and two workshops associated with antiretroviral clinical pharmacology (pharmacokinetics - drug absorption, distribution, metabolism, and elimination; and pharmacodynamics - drug effects). This report will summarize pertinent data from these presentations and posters. The reviews have been organized into the following sections: - Intracellular nucleoside analog triphosphates (RBV & NRTIs)
- Pharmacogenetics and drug transporters
- Non-nucleoside reverse transcriptase inhibitors (EFV PK): statins; sex & race; TDF and hepatic disease
- Protease inhibitors (triple PI combinations)
- Antiretroviral pharmacology relevant to treatment in resource poor settings
- Novel antiretrovirals
PHARMACOLOGY OF INTRACELLULAR NUCLEOSIDE ANALOG TRIPHOSPHATES
Nucleoside analogs (NA) are pro-drugs; the drug itself is inactive and must become phosphorylated (PO4 groups are chemically attached) inside the cell three times in order to be pharmacologically active. Measuring intracellular NA-triphosphates is challenging since concentrations are very low and many cells (e.g. 2 to 10 million peripheral blood mononuclear cells(PBMCs)) are needed for the analysis. Nevertheless, it is necessary to study intracellular NA-triphosphates in order to characterize and understand their clinical pharmacology.
Zidovudine converting to d4T-triphosphate in patients?
Melendez et al (abstract 597) attempted to replicate the recent AIDS report of zidovudine-triphosphate (ZDV-TP) chemically converting to stavudine-triphosphate (d4T-TP) in HIV-infected patients treated with ZDV. (Becher, et al. AIDS 2003; 17:555-61) A different analytical method was used in this study, but the two methods were similarly sensitive for ZDV-TP and d4T-TP. In this study, 528 samples were analyzed for d4T-TP from patients on ZDV. d4T-TP was not detected in any patient sample despite a robust signal for ZDV-TP. In addition, a cell line (CEM) was incubated with varying concentrations of ZDV and 20 such samples were analyzed for d4T-TP. Again, no d4T-TP was detected despite robust signals for ZDV-TP. These findings from an experienced group of analytical chemists throw into question whether or not ZDV-TP converts to d4T-TP in patients.
Effect of drug transporters on NA-triphosphates
Rodman and colleagues (abstract 598) also examined ZDV phosphates (mono-,di-, and tri) and how they are affected by multidrug-resistance protein 4 (MRP4) expression. MRP4 is a human cell membrane transporter that pumps NA-monophosphates out of the cell interior, which would presumably reduce the pharmacologically active triphosphate concentrations. The investigators incubated equal amounts of ZDV in a CEM cell line that highly expressed MRP4 versus a CEM cell line that did not. They found approximately 5 to 9-fold more ZDV-phosphates (including triphosphate) in the cells that did not highly express MRP4. An MRP4 inhibitor (MK571) increased the triphosphate concentrations 2-fold in the MRP4 expressing cells and also 1.5-fold in the cells that did not highly express MRP4 (these cells express lesser, but detectable amounts of MRP4). Additionally, the half-life of the intracellular ZDV-TP was twice as long in the cells that did not highly express MRP4. These experiments highlight MRP4 as a potential source of patient-to-patient variability in intracellular NA triphosphates. This should be investigated further.
Triphosphate studies with a new nucleoside analog (SPD754)
Two posters described the intracellular triphosphate pharmacokinetics of SPD754, an investigational cytosine analog, in HIV-infected patients. Adams and colleagues (abstract 599) determined that triphosphate concentrations were proportional with dose and were related to SPD754 plasma concentrations. Furthermore, SPD754-triphosphate concentrations were similar in magnitude to triphosphate concentrations in patients for lamivudine (3TC), another cytosine analog. The half-life was approximately 6 to 7 hours, which, at first glance, is more conducive to twice-daily dosing compared with once a day. Bethell and colleagues investigated possible intracellular interactions between 3TC-triphosphate and SPD754-triphosphate in healthy volunteers, since these drugs utilize the same enzymes for phosphorylation (abstract 138). There was no drug-drug interaction between these agents in terms of plasma concentrations. However, the intracellular concentrations of SPD754-triphosphate were reduced 6-fold. 3TC-triphosphate concentrations were not altered. This study demonstrated the importance of conducting drug-drug interaction studies for intracellular nucleoside analog triphosphates, particularly when there is suspicion of an interaction.
Ribavirin interactions with NA-triphosphates
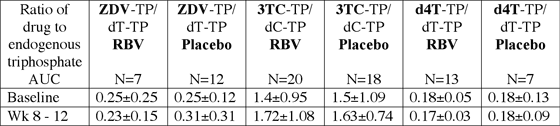
Two studies examined intracellular drug interactions with NA and agents used for hepatitis treatment, specifically pegylated-interferon (PEG-IFN) and ribavirin (RBV). These data are important since in vitro studies indicate that RBV increases intracellular production of ddA-triphosphate, the active moiety for didanosine (ddI), but inhibits production of intracellular ZDV and d4T triphosphates. Gries, and colleagues (abstract 136 LB) described ZDV, 3TC, and d4T-triphosphates in HIV-HCV co-infected patients before and after starting RBV (800 mg /day) or placebo plus PEG-IFN 180 mcg/wk. The area under the concentration time curve (AUC) was calculated for ZDV, d4T, and 3TC-triphosphates and the endogenous counterparts, deoxythymidine- (dT-TP) and deoxycytidine-triphosphates (dC-TP), respectively. The ratio was then calculated for the AUCs of the drug-triphosphates/endogenous-triphosphates. These results are displayed in the table below. There were no significant differences in the ratios of any combination when combined with RBV or placebo. The authors also measured ZDV, d4T and 3TC plasma concentrations and found no significant plasma interactions with RBV or placebo. They concluded that RBV does not interfere with the plasma or intracellular-triphosphate disposition of ZDV, d4T, and 3TC. In contrast, Hennessy and colleagues (abstract 822) found a 44% reduction in the ZDV-TP/dT-TP AUC ratio (P=0.02) 14 days after initiating HCV therapy in 6 patients receiving PEG-IFN and RBV (1000mg to 1200mg/day). This resulted from a 1.8-fold increase in ZDV-TP, but a larger increase in dT-TP AUC (3-fold). The d4T-TP/dT-TP AUC ratio did not change in the 5 patients studied, whereas the 3TC-TP/dC-TP AUC ratio increased transiently at day 3 in 9 patients and was attributed to a 1.4-fold increase in the 3TC-TP AUC. Although it is unclear why the results of these two studies differ, higher RBV doses and earlier sampling after initiation of HCV therapy in the second study may contribute to this disparity. These two abstracts provide evidence that there is not a major intracellular interaction among these agents, which is generally consistent with clinical studies that do not suggest reduced efficacy of ZDV, d4T, and 3TC with HCV therapy.
PHARMACOGENETICS AND DRUG TRANSPORTERS
Pharmacogenetics refers to the study of the inherited (genetic) basis for patient-to patient differences in drug response. Genetic differences have been identified in cytochrome P450 (CYP) enzymes, which metabolize many drugs and in P-glycoprotein (P-gp), which is a transporter involved in drug clearance. HIV protease inhibitors (PIs) are known substrates for P-gp and CYP; thus, studying these potential genetic differences may predict PI activity.
Pharmacogenetics of indinavir exposures and responses
Anderson and colleagues (abstract 619) described relationships between antiviral responses and indinavir (IDV) oral clearance (CL) with CYP 3A5 and P-gp (MDR1 gene) genotypes in 33 antiretroviral-nave patients initiating ZDV, 3TC, and IDV. The presence of a certain CYP 3A5 genotype indicates whether or not a person expresses the CYP 3A5 drug-metabolizing enzyme. IDV is partly metabolized by CYP 3A5. In this study, 11 subjects (including 7 of 7 African-Americans) expressed CYP 3A5 and 22 did not. IDV CL was 43% higher in CYP 3A5 expressors (blood levels would be lower) compared with those who did not express CYP 3A5 (P=0.002). This relationship held when adjusted for African-American race and P-gp genotypes. There was also a relationship between the reduction in HIV-RNA from baseline to week 52 (or study exit) and P-gp genotypes GT or TT versus GG at position 2677 in the MDR1 gene. Those with GT or TT had a 3.3 log reduction whereas those with GG had a 2.2 log reduction (P=0.003). This relationship remained significant when adjusted for baseline HIV-RNA, CYP 3A5 expression, IDV oral clearance, and African-American race. This study indicated that CYP 3A5 expression, which is more common in blacks than whites, may be associated with risk of low IDV blood concentrations. P-gp genotypes may be associated with antiviral responses.
Molecular basis of drug-drug interactions caused by PIs
Induction of CYP enzymes and transporters occurs when their genes are transcriptionally activated by pregnane X receptor (PXR), constitutive androstane receptor (CAR) or farnesoid X receptor (FXR). Marzolini and colleagues investigated these molecular events (abstract 135). The PIs ritonavir (RTV), lopinavir (LPV), amprenavir (APV) and nelfinavir (NFV) were all partial PXR agonists. In other words, these agents activated this receptor, but not completely. The combination of LPV + RTV + APV showed a synergistic effect on PXR, which is consistent with the drug-drug interaction seen with this combination clinically. NFV and RTV activated CAR, and only NFV activated FXR. Experiments were confirmed by measuring the increase of CYP 3A proteins in primary human liver cells. These studies provide a molecular basis for the unpredictable drug-drug/herbal interactions with PIs observed in patients (see pharmacology of protease inhibitors below).
P-gp and drug concentrations and responses
P-gp activity on CD4 cells and MDR1 (P-gp) genotype in 205 HIV-infected patients was examined by Hulgan and colleagues (abstract 617). A total of 148 (72%) patients were taking antiretroviral therapy, which included a PI in 78 (38%). P-gp activity was inversely correlated with plasma HIV-RNA and among PI treated patients, those with a TT genotype at 3435 had 38% less P-gp activity than those with CC at 3435 in the MDR1 gene (P=0.04). This study indicates that increased P-gp activity is correlated with less plasma HIV-RNA and a potential interaction may exist among PIs, P-gp activity, and the genotype at 3435 in the MDR1 gene. Chandler and colleagues (abstract 618) also examined P-gp and its relationship with the expression of CXCR4, which is a co-receptor used by HIV-1 for entry into cells. Phytohemagglutinin (PHA)-stimulated peripheral blood mononuclear cells (PBMCs) were examined and infected with HIV ex vivo. The P-gp inhibitor XR9576 was added to some of the cultures. The concentration of saquinavir (SQV) required to inhibit viral replication by 50% (SQV IC50) was also determined. Correlations between IC50, P-gp and CXCR4 were assessed. Results indicated that P-gp and CXCR4 expression were highly correlated (rho=0.52; P<0.001). P-gp and p24 recovery were weakly correlated (P=0.06). XR9576 did not influence p24 recovery. The SQV IC50 was positively correlated with both the P-gp and CXCR4 expression; P=0.003 and P=0.03, respectively. These in vitro studies suggest co-regulation of P-gp and CXCR4. The activity of SQV was associated with the expression level of these proteins.
Owen and colleagues (abstract 619b) studied P-gp (MDR1 gene) genotypes in a heterogeneous population of experienced and nave patients initiating SQV/RTV (1000mg/100mg) bid or IDV/RTV (800mg/100mg) bid. Random drug concentrations were collected and trough concentrations were estimated on the basis of these concentrations. There were no relationships between MDR1 genotypes and PI drug concentrations. The 2677 GG patients had lower baseline CD4 cells than the TT group. In addition, whereas the GG group had no increase in CD4 cells during follow up, the TT group had a 70 cell change (P=0.04). van der Ende and colleagues (abstract 605) investigated the CYP 3A5 expression, CYP 3A4 *1B, and P-gp (MDR1) genotypes in 186 clinic patients using NVP who were also undergoing routine therapeutic drug monitoring. No relationships were found between these genotypes and high (>8.5 mcg/mL; n=19) or low (<3.8 mcg/mL; n=20) NVP concentrations. Subjects with high NVP concentrations tended to be non-Caucasian and had positive hepatitis serology.
The pharmacology of protease inhibitors with regard to P-gp remains an area of confusion. In future studies, potential sources of variability should be controlled as much as possible.
PHARMACOLOGY OF NON-NUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS (NNRTI)
The oral session on pharmacology and new antiretroviral agents began with several presentations discussing the relationships between efavirenz (EFV) pharmacokinetics, nNRTI resistance, side effects and race.
The PK of Efavirenz after treatment interruption
Taylor and colleagues (abstract 131) began the oral session by reporting on the presence of EFV in plasma up to 3 weeks after drug discontinuation. Plasma samples from 10 HIV-infected patients were analyzed for EFV concentrations at baseline and at days 4, 7, 14 and 21 after stopping EFV (background therapy was continued). At baseline, the median EFV concentration was 3004 ng/mL compared with 149 ng/mL at day 14. Three patients had EFV concentrations >1000 ng/mL 2 weeks after stopping therapy. Although the estimated therapeutic range for EFV is 1100-4000 ng/mL, EFV plasma concentrations between the range of 100 and 1000 ng/mL may be sufficient to impart selective pressure for nNRTI resistance. The persistence of EFV in plasma in these subjects 2 weeks after stopping therapy raises the question of how to manage this time of potential nNRTI monotherapy. Perhaps NRTI therapy should continue for 7 to 14 days after stopping nNRTI therapy, or perhaps nNRTI therapy should be changed to a PI approximately 1 to 2 weeks prior to stopping all antiretrovirals. These research questions should be addressed.
The PK of Efavirenz according to sex and race
Two ACTG studies evaluated relationships among EFV PK, covariates associated with PK, CNS side effects, and antiviral effects in HIV-infected, naive patients. H. Ribaudo and colleagues (abstract 132) used a population pharmacokinetic (PK) model to obtain estimates of EFV CL (inversely proportional to AUC). The aim of the study was to examine associations between these PK parameters with the weight, sex and race of study subjects. Plasma samples from 190 subjects were available for analysis, 19% from women. Race distribution was 53% white, 32% black and 15% Hispanic. Results indicated a 32% increase in CL among white, non-Hispanic subjects (corresponds with lower AUC) compared with black and Hispanic subjects (p<0.001). No associations were noted with gender (p>0.26). During study follow-up, 31 patients discontinued EFV, primarily due to CNS symptoms (n=5). There was a trend towards an increased rate of EFV discontinuation associated with decreased CL (corresponds with higher AUC) (p=0.052) and increased Cmax (p=0.048). Haas and colleagues (abstract 133) further analyzed data from this patient population by examining the genetic variation in CYP2B6, the CYP responsible for the metabolism of EFV. The genetic sequences of CYP 3A4/3A5 and P-gp were also analyzed. Briefly, the authors found that a homozygous T/T genotype at position G516T was more common in blacks (20%) than whites (3%) and was associated with a 3-fold higher AUC than subjects with G/G (P=0.001). In a multivariate analysis, the only variables associated with EFV AUC were the CYB 2B6 G516T genotype and, to a much lesser extent, CYP 3A4/3A5 genotypes. Finally, there were significantly more CNS side effects in the CYP 2B6 TT group (P=0.04), but there were no relationships with virologic/immunologic outcomes.
|
|
| |
| |
 |
|
| |
| |
In contrast to data presented from these ACTG studies, abstract 604 reported that race had no effect on EFV systemic exposure, as determined by AUC. Women, however, had lower systemic exposure to EFV compared with men and the sex difference persisted after adjustment for racial group, weight and co-administered medications. This retrospective analysis of PK data from 6 ACTG studies also examined the effect of sex, race and weight on IDV (all IDV was in dual-PI regimens), NFV and NFVs active metabolite, M8. There were no significant sex differences in systemic exposure of the PIs; however, African American subjects had significantly higher IDV AUC and lower NFV and M8 AUC.
Drug-drug interaction between efavirenz and statins
A drug-drug interaction study was conducted between EFV and the lipid lowering agents, simvastatin (SIM) and atorvastatin (ATR) in healthy volunteers (abstract 603). Certain ARVs have been associated with increased serum LDL-cholesterol and triglycerides, and many patients require statins to lower these levels. SIM and ATR are metabolized in the liver by CYP450 3A4 enzymes, which can be induced or inhibited by EFV, and altered plasma concentrations of statins could cause serious side effects (myalgias and rhabdomyolosis), or decreased anti-lipid responses. Gerber and colleagues found that EFV induced the metabolism of SIM and ATR and/or their active metabolites by 60% and 50%, respectively. Neither SIM nor ATR appeared to affect the plasma exposure of EFV. These findings suggest that EFV may decrease the overall anti-lipid response during therapy. However, statin dose escalation will require increased monitoring for toxicity and efficacy.
Drug-drug interaction study between TDF and d4T
Abstract 602 also evaluated the drug-drug interaction between extended release stavudine (d4T XR) and tenofovir (TDF). Both antiretrovirals are potential components of once-daily HAART regimens and TDF has been shown to increase plasma concentrations of other NAs, such as ddI. An earlier study demonstrated that TDF had no significant effect on the PK of d4T XR (Kaul S, et al. 10th CROI, abstract 534-W). The current study evaluated TDF PK in 18 healthy subjects receiving TDF 300 mg once daily for 8 days with a subsequent PK followed by a single dose of d4T XR 100 mg on day 9 with a subsequent PK. Once daily d4T XR had no significant effect on the PK of TDF suggesting that no dose adjustments are required when these 2 antiretrovirals are administered concomitantly.
Hepatic disease and TDF PK and TDF effects on adefovir and ribavirin
TNF PK were also evaluated in patients with hepatic impairment (abstract 600). A single dose of TDF 300 mg was administered to otherwise healthy subjects and subjects with moderate or severe hepatic impairment; mean Child-Pugh scores of 8 and 10.8, respectively. The AUC and Cmax of TDF were increased approximately 1.35-fold between the healthy subjects and those with severe hepatic impairment. The values for the moderate impairment group were between these groups. However, these differences were not deemed to be significant by the authors and dose adjustments in patients with hepatic impairment were not recommended. The PK of adefovir (ADV) and ribavirin (RBV), two drugs commonly used in patients with hepatitis, were also examined when administered with TDF in healthy subjects. Briefly, a single dose of ADV 10 mg was with subsequent PK. This was followed with multiple doses of TDF 300 mg and another subsequent ADV PK. This was repeated in another group and a single dose of RBV 600 mg, followed with multiple TDF 300 mg doses with similar RBV PK sampling. The PK of ADV and RBV were not significantly different when dosed with or without TDF.
PHARMACOLOGY OF PROTEASE INHIBITORS
Intracellular SQV and RTV
Ford and colleagues (abstract 601) determined plasma and intracellular (cell-associated) concentrations and PK of SQV (hard-gelatin formulation)/RTV 1600/100 mg once daily in 12 HIV-positive patients receiving this combination. Plasma and PBMCs were collected at steady-state at 2, 6, 12, and 24 hours post-dose. Relationships between SQV and RTV accumulation in lymphocytes and lymphocyte subsets (CD4+, CD8+, and CD56+ cells) and P-gp expression on these lymphocytes were also examined. The median accumulation ratio (cellular AUC0-24h/ plasma AUC0-24h) of SQV and RTV was 3.31 and 1.46, respectively, indicating that cell-associated SQV and RTV are 3.3 and 1.5-times higher than in plasma. The cell-associated half-lifes of SQV and RTV were longer than in plasma (5.9 versus 4.5 hours for SQV (p=0.034) and 10.4 versus 7.5 hours for RTV (p=0.033)). The authors hypothesized that relatively higher intracellular accumulation of SQV trough concentrations may allow greater forgiveness for missed or late doses with this once daily combination. No relationship was observed between SQV and RTV accumulation and P-gp expression on total lymphocytes or CD4+, CD8+, or CD56+ lymphocyte cell subsets.
Evaluation of ATV inhibitory quotient (IQ)
Barrios and colleagues (abstract 606) assessed the predictive value of baseline drug resistance genotypes, atazanavir (ATV) plasma trough levels at week 12, and genotypic inhibitory quotients (GIQ = ATV trough (or estimated trough) measured at week 12/number of protease resistance mutations (PRM) at baseline) on virologic response at week 24 in a group of patients taking ATV 400 mg once daily along with two additional drugs as part of an ATV expanded access program. Seventy-eight percent of patients received TDF. None of these patients received RTV along with ATV (TDF has been shown to reduce ATV AUC and Cmin.) Data were analyzed from 92 and 52 patients followed for 12 and 24 weeks, respectively. At week 12, in 43 patients, median (IQR) estimated ATV Cmin was 0.14 mg/mL (0.06-0.26 mg/mL) in responders (1 log drop in HIV RNA or <50 copies/mL at week 24) and 0.12 mg/mL (0.05-0.22 mg/mL) in non-responders. In contrast to other studies, the median estimated week 12 ATV Cmin was similar in patients who were and were not taking TDF (0.11 mg/mL and 0.12 mg/mL, respectively) however, the number of patients in each group was not specified. In multivariate analysis, virologic response at week 24 was not associated with estimated week 12 ATV Cmin or number of baseline PRM. The median (IQR) number of baseline PRM in the study population was 2 (1.5-5) and tended to be lower in responders versus non-responders. The authors did report that a higher GIQ was associated with a greater drop in HIV RNA at week 24, suggesting that GIQ in this study is primarily influenced by number of baseline PMR.
PK of triple PIs: ATV/SQV/RTV
Boffito and colleagues (abstract 607) evaluated the short-term safety and steady-state PK of ATV/SQV (hard-gelatin formulation)/RTV 300/1600/100 mg once daily in 18 HIV-infected subjects. PI concentrations were measured over 24 hours at baseline for SQV/RTV 1600/100 mg once daily and ten days later for ATV/SQV/RTV. Subjects took all study medications with a high fat (20 g) meal to improve absorption. Geometric mean ratios (GMR) of plasma Ctrough, Cmax, AUC0-24h, and half-life for SQV and RTV were compared before and after the addition of ATV. There was a statistically significant increase in the GMR of all four metrics for SQV following the addition of ATV indicating that ATV improved SQV exposure and prolonged half-life. The geometric mean (95% CI) SQV Ctrough before and after addition of ATV was 87 ng/mL (72-139 ng/mL) and 184 ng/mL (140-311 ng/mL), respectively. For RTV, only the GMR of Cmax and AUC0-24h was significantly increased. Interestingly, the GMR of the RTV half-life was significantly decreased.. Geometric mean ATV plasma Ctrough, Cmax, AUC0-24h, and half-life were 767 ng/mL, 4982 ng/mL, 51,036 ng*h/mL and 9.8 hours, respectively. Of note, following the addition of ATV in this study, 6/18 (33%) subjects developed clinically relevant hyperbilirubinemia: scleral icterus (n=4), jaundice (n=2). The authors concluded that this combination should be evaluated further.
PK of triple PIs: FPV/SQV/RTV
In another study by Boffito and colleagues (abstract 608) the short-term safety and steady-state PK of SQV (hard-gelatin formulation)/fosamprenavir (FPV, APV prodrug) 1000/700 mg twice daily along with 100 mg or 200 mg of RTV were evaluated in 18 HIV-infected subjects. PI concentrations were measured over 12 hours on day 1 for SQV/RTV 1000/100 mg twice daily, on day 11 for SQV/RTV/908 1000/100/700 mg twice daily, and on day 22 for SQV/RTV/908 1000/200/700 mg twice daily. At each visit, study medications were taken with a high fat (20 g) meal to improve absorption. Administration of 908 700 mg twice daily along with SQV/RTV 1000/100 mg twice daily lead to statistically non-significant decreases in geometric mean SQV Ctrough, Cmax, AUC0-12h, and half-life of 24%, 9%, 14%, and 13%, respectively. However, statistically significant decreases were seen in geometric mean RTV Ctrough, Cmax, AUC0-12h, and half-life of 54%, 29%, 40%, and 24%, respectively. The authors speculate that the decrease in RTV Ctrough may have contributed to lower SQV concentrations in this treatment arm. Increasing the dose of RTV to 200 mg twice daily compensated for reductions in both SQV and RTV. The PK of APV following 908 was unchanged in the presence of RTV 100 mg or 200 mg twice daily. Based on these data, the authors conclude that optimal dosing of this PI combination appears to be SQV/RTV/FPV 1000/200/700 mg twice daily.
Dose-management of triple PIs: APV/LPV/RTV
Wynn Vezina and colleagues (abstract 609) described pharmacologic management of the drug-drug interaction between LPV/RTV and APV in 12 HIV-infected subjects starting salvage therapy with LPV/RTV/APV 400/100/600 mg twice daily in combination with NRTIs (including TDF). LPV and APV concentrations were measured over 12 hours after 2 weeks and random concentrations were measured monthly out to 24 weeks. Median LPV C12h and AUC0-12h were 3- and 2-fold lower, respectively, than the manufacturers reported mean Ctrough (5500 ng/mL) and AUC0-12h (82,800 h*ng/mL). Median LPV oral clearance (CL/F) was higher than the 6-7 L/h reported in the package insert. Median APV C12h and AUC0-12h were 4- and 1-fold higher, respectively, than the manufacturers reported mean Cmin (280 ng/mL) and AUC0-12h (18,500 h*ng/mL) and similar to values reported in the literature for APV/RTV 600/100 mg twice daily. Measured LPV C12h <3000 ng/mL and/or APV C12h <1000 ng/mL were threshold values used to guide recommendations for dose adjustments. 6/12 (50%) subjects had LPV/RTV dose increases (533/133 mg n=2; 666/166 mg n=4) and 3/12 (25%) subjects had APV dose increases (750 mg n=2; 900 mg n=1). In the 6 subjects with LPV/RTV dose increases, the pre-dose adjustment median (range) C12h was 960 ng/mL (833-2943 ng/mL). Following dose adjustments, the predicted median (range) C12h (ng/mL) increased to 4465 ng/mL (1752-7841 ng/mL). One subject had LPV/RTV decreased to 267/67 mg twice daily secondary to diarrhea and LPV C12h went from 9966 to 3978 ng/mL. In this study, no adverse events resulted from PI dose increases. The authors concluded that the magnitude of the interaction between LPV/RTV and APV is difficult to predict and optimal management may require pharmacokinetically-guided dose individualization.
Attempted dose-management of FPV/LPV/RTV
Corbett and colleagues (abstract 611) evaluated whether physical separation of LPV/RTV and 908 could overcome the drug-drug interaction between these two PIs (exposure of both PIs is decreased 48-69% during co-administration). Eleven HIV negative volunteers completed this prospective, randomized, 3-treatment, 3-period, 3-way crossover study. All subjects received 908/LPV/RTV 700/400/100 mg twice daily for 10 days and then started either 908/LPV/RTV 700/400/100 mg taken simultaneously twice daily for 7 days, 908/RTV 700/100 mg followed 4 hours later by LPV/RTV 400/100 mg each taken twice daily for 7 days, or 908/RTV 1400/200 mg followed 12 hours later by LPV/RTV 800/200 mg each taken once daily for 7 days. All 11 subjects received all 3 treatments according to randomization. During simultaneous administration, geometric mean data for APV Cmax,ss, AUCl,ss, and Cl,ss were 1.88 mg/mL, 10.21 mg*h/mL, and 0.52 mg/mL, respectively, compared with historic PK data for 908/RTV 700/100 mg twice daily of 6.73 mg/mL, 41.97 mg*h/mL, and 2.34 mg/mL, respectively. Similarly, geometric mean data for LPV Cmax,ss, AUCl,ss, and Cl,ss were 8.10 mg/mL, 69.50 mg*h/mL, and 2.98 mg/mL, respectively, compared with historic PK data for LPV/RTV 400/100 mg twice daily of 10.32 mg/mL, 93.25 mg*h/mL, and 5.85 mg/mL, respectively. Separating the PIs by 4 or 12 hours did not improve the PK of APV, but did correct the PK of LPV. The authors concluded that further investigation is needed to determine optimal dosing of this PI combination.
Another attempted dose-management of FPV/LPV/RTV
FPVis fosamprenavir, aka 908. Wire and colleagues (abstract 612) assessed two alternative dosing strategies in healthy volunteers in an attempt to overcome the drug-drug interaction between LPV/RTV and 908. At steady state, 908/LPV/RTV 1400/533/133 mg twice daily was compared with standard LPV/RTV 400/100 mg and standard 908/RTV 700/100 mg twice daily (APV10011). A companion study investigated 908/RTV/LPV/RTV 700/100/400/100 mg twice daily versus the standard regimens (APV10012). After a 28-day washout between treatments, subjects in both studies switched to the alternative regimen for another 14 days. Of 36 patients, thirteen subjects in APV10011 and 16 subjects in APV10012 prematurely discontinued, primarily due to adverse events (gastrointestinal and rash). In 13 subjects who completed APV10011, geometric mean APV Cmax,ss, AUCl,ss, and Cl,ss decreased 13%, 25%, and 42% respectively, for the regimen 908/LPV/RTV compared with 908/RTV alone. In 10 subjects who completed APV10012, geometric mean APV Cmax,ss, AUCl,ss, and Cl,ss decreased 59%, 64%, and 66% respectively, for the regimen 908/RTV/LPV/RTV compared with 908/RTV alone. For LPV, the authors reported that increased doses of LPV/RTV or administering additional RTV maintained or increased LPV concentrations in the presence of 908. As in abstract 611, the authors concluded that optimal dosing recommendations couldnt be made for the combination of FPV and LPV/RTV.
Lopinavir concentrations and lipid changes
Best and colleagues (abstract 613) explored relationships between LPV exposure and change in lipids in subjects taking part in on ongoing adherence and therapeutic drug monitoring clinical trial (CCTG 578). In 37 subjects, after 2 weeks of LPV/RTV, LPV and RTV plasma samples were collected at baseline, 2, and 4 hours post-dose. Post-hoc Bayesian estimates of individual subjects LPV and RTV concentrations at 2 (C2), 4 (C4), and 12 (C12) hours after a dose were determined using a nonlinear mixed effects modeling (NONMEM) approach. Mean fasting total cholesterol (TC), high-density lipoprotein (HDL), low-density lipoprotein (LDL) and triglycerides (TG) increased 22%, 17%, 10%, and 38%, respectively, from baseline to week 24 of the study. No subjects were taking lipid-lowering agents. Neither LPV nor RTV concentrations were associated with changes in HDL, LDL, and TG at week 24. Increasing LPV C2 and C4 were associated with lower changes in TC at week 24 (p0.02). LPV C12 and RTV C2 and C4 showed similar trends, but were less predictive. In another study, Gonzalez de Requena and colleagues (abstract 610) found that LPV and RTV Ctrough at month 3 in subjects taking LPV/RTV as part of salvage therapy was weakly correlated with percent increase in TC and TG from baseline to week 48 (r=0.28, p=0.04 for LPV; r=0.314, p=0.018 for RTV).
Counteracting efavirenz induction of amprenavir with other PIs
Morse and colleagues (abstract 614) compared the PK of APV when taken alone, with EFV and with a second PI, either NFV, IDV, RTV, or SQV, in a population of HIV negative volunteers. All PK studies were conducted at steady-state. Subjects were randomized to receive APV 600 mg every 12 hours + EFV 600 mg every 24 hours (Arm A), Arm A + NFV 1250 mg every 12 hours (Arm B), Arm A + IDV 1200 mg every 12 hours (Arm C); Arm A + RTV 100 mg every 12 hours (Arm D), or Arm A + SQV 1600 mg every 12 hours (Arm E). A stepwise introduction of EFV and the second PI was used in this study. Each subject underwent three PK visits (day 0 on APV alone, day 14 on APV + EFV, and day 21 on APV + EFV + second PI). APV clearance increased during the 10-day period of combined EFV indicating that EFV increased the metabolism of APV and reduced APV plasma concentrations. Adding a second PI had variable effects on APV exposure. The median percent change in APV GMR of AUCs from day 14 to day 21, was 22% (A), 316% (B), 293% (C), 1068% (D), and 20% (E), indicating that of the second PIs added, NFV, IDV and RTV increased APV AUC in the presence of EFV. SQV had a minimal effect on APV exposure. The authors concluded that NFV and IDV may be alternatives to RTV for PK boosting of APV.
Lopinavir concentrations and lipid changes
Bertz and colleagues (oral abstract 134) assessed predictors of response to high-dose LPV/RTV. Thirty-three multi-drug experienced HIV-infected subjects were randomized to receive LPV/RTV 666/167 mg or LPV/RTV 400/300 mg, both twice daily along with two to three NRTIs (including TDF). Plasma samples for LPV PK were obtained over 12 hours at week 3. The median (range) LPV fold change in IC50 was 4.1 (0.6 to 273). LPV Ctrough values were similar for both regimens, 60-70% higher compared with standard dose. The median (range) LPV IQ was 27 (0.7 to 438). By week 48, 64% of subjects had HIV RNA <400 copies/mL and 48% had HIV RNA <50 copies/mL. In an adjusted multiple logistic regression analysis, lower baseline HIV RNA, higher LPV IQ and the number of active NRTIs in a subjects treatment regimen were associated with achieving HIV RNA <400 copies/mL. The authors concluded that higher doses of LPV/RTV may have provided LPV concentrations that were sufficient to overcome certain degrees of reduced LPV phenotypic susceptibility. Of mention, in abstract 610, a multivariate analysis of data from 65/126 subjects who received 48 weeks of salvage therapy that included LPV/RTV 400/100 mg twice daily, found that both LPV Ctrough >5 mg/mL (measured at month 3) and 6 baseline protease resistance mutations were independent predictors of virologic response (>1 log decrease in HIV RNA and/or <50 copies/mL) at week 48. This study further highlights how higher LPV concentrations and fewer baseline protease resistance mutations may contribute to improved response to LPV/RTV-based salvage therapy.
ANTIRETROVIRAL PHARMACOLOGY RELEVANT TO TREATMENT IN RESOURCE POOR SETTINGS
Several pharmacology abstracts presented at this years conference have the largest impact on HIV treatment in resource poor settings. Penzak and colleagues (abstract 581) analyzed 6 ARV medications from 6 manufacturers and 4 international sources. With the exception of RTV, which was not stored under continual refrigeration, the active ingredient in each of the drug products was within USP specifications of 88 and 115% of labeled amounts. Similar results were reported by Ramachandran and colleagues (abstract 582) who evaluated the drug content of EFV, NVP, ZDV, d4T and 3TC formulations manufactured in India. The mean drug content of all preparations compared well with the proprietary formulations ( 5%) and variability ranged from 0.01 to 8.3%. Zala and colleagues (abstract 615) compared the pharmacokinetics of RTV boosted generic (Inhibisam) versus brand (Crixivan) IDV. Ten HIV-infected patients on stable antiretroviral therapy participated in a prospective cross-over study to receive either Crixivan or Inhibisam 800 mg and RTV 100 mg bid. Pharmacokinetics analysis was performed on day 1 and again 14 days after the crossover. Cmax, Cmin and AUC for Inhibisam and Crixivan were 7531 and 7896 ng/mL, 1383 and 1007 ng/mL and 50630 and 42635 ng*h/mL, respectively. The authors concluded that IDV exposure was not statistically different between the two formulations. The pharmacokinetics of IDV/RTV at doses of 400/100 mg every 12 hours were reported (abstract 616). Results from this abstract could be attractive for treatment in resource poor settings since the reduced dose of IDV/RTV is the least expensive boosted PI combination. Boyd and colleagues reported the PK parameters for IDV/RTV 400/100 mg bid in 19 treatment nave, HIV-infected patients. Three patients had a Cmin <0.1 mg/L, which was the target value and virologic failure occurred in 1 patient. 16 patients had a Cmin >0.1 mg/L and virological failure occurred in 1 patient. The authors concluded that a therapeutic Cmin was achieved in >78% of subjects and the short-term virologic response was satisfactory. However, it is not optimal for 3 of 16 (19%) of subjects to have IDV concentrations below the target.
NOVEL ANTIRETROVIRALS: new CCR5 inhibitor (GW873140)
Piscitelli and colleagues (abstract 139) presented the safety, tolerability, and pharmacokinetics of a new CCR5 receptor inhibitor, GW873140 in 70 healthy volunteers. Pharmacokinetics were determined after escalating single doses then again after multiple doses. Mild and moderate GI cramping, nausea, and diarrhea were observed and some CNS symptoms were reported. No ECG alterations were found. Rare increases in ALT/AST, CPK, lipase, and cholesterol were observed, but it was not clear these were drug related. The pharmacokinetics were fairly dose-proportional. Food increased drug exposure by about 2-fold. Twenty-four hours post dose, GW873140 blood levels were near or below the detection limit of the assay (1 ng/mL). However, at this same time, about 70% of the CCR5 receptors were occupied by the drug, which suggests the potential for once-daily dosing. This study supports further pursuit of this agent in HIV-infected patients.
|
|
| |
| |
|
| |
|
|
|
|
|