| |
Prevention of (HBV) Hepatocellular Carcinoma
|
| |
| |
EDITORIAL
New England Jnl of Medicine Oct 7, 2004 Vol 351:1567-1570
Jack R. Wands, M.D.
From the Liver Research Center and Brown Medical School — both in Providence, R.I. Hepatocellular carcinoma is a significant problem worldwide, and the incidence is increasing in the United States.1 Approximately 90 to 95 percent of these tumors result from the biologic consequences of persistent hepatitis B virus (HBV) and hepatitis C virus (HCV) infection. There are about 450 million chronic carriers of HBV and 200 million carriers of HCV. The general clinical sequence of events is exposure to HBV early in life, and then chronic hepatitis, liver injury and regeneration, development of fibrosis, and cirrhosis. The risk of the development of hepatocellular carcinoma is about 25 to 35 times as high among patients with chronic HBV infection as it is among those without the infection, but it is 130 times as high among patients with both HBV and HCV infection as it is among uninfected persons.2 The annual number of deaths from HBV-related liver disease throughout the world is about 1.2 million.
HBV is the prototype member of the Hepadnaviridae family, which also infects ducks, ground squirrels, and woodchucks. These small, partially double-stranded DNA viruses contain four overlapping genes that encode for the nucleocapsid, envelope, polymerase with reverse-transcriptase activity, and X proteins.
Most of our knowledge about HBV replication is derived from the study of duck HBV, which can be grown in primary hepatocyte culture. Woodchuck HBV was discovered in animals that were dying from hepatocellular carcinoma at the Philadelphia Zoo. Hepatocellular carcinoma develops within a four-year period in almost all eastern woodchuck pups exposed to woodchuck HBV at birth. Thus, the duck and the woodchuck have been important animal models for the development of anti-HBV agents and have revealed fundamental genetic events involved in hepatocarcinogenesis.
Some of the molecular and cellular mechanisms3 associated with the development of HBV-related hepatocellular carcinoma are shown in Figure 1. These mechanisms include chronic liver injury and increased cell turnover, which confer a predisposition to hepatocyte transformation; the integration of HBV DNA into the DNA of the hepatocyte and the promotion of genomic instability by HBV DNA; expression of the HBx protein (which has transcriptional transactivator activity and up-regulates growth-promoting genes) during persistent viral infection; a high HBV-replication phenotype and naturally occurring mutations in the core promoter region, which have been identified as viral risk factors for the development of hepatocellular carcinoma; and progression of chronic liver disease to severe fibrosis, which greatly increases the risk of hepatocellular carcinoma, since approximately 75 to 85 percent of such tumors develop in the cirrhotic liver. Other known major risk factors include exposure to aflatoxins in some areas of the world and excessive alcohol consumption.3 With the development of diagnostic techniques sensitive enough to detect very low levels of serum HBV DNA (<100 copies per milliliter), it has become increasingly apparent that many patients with chronic HCV infection are also infected with low levels of HBV. In this setting, HBV maintains its oncogenic properties,5 and evidence is accumulating that occult HBV infection (defined as <10,000 virions per milliliter of serum) may be associated with chronic hepatitis and cirrhosis of heretofore unknown cause.6
|
|
| |
| |
 |
|
| |
| |
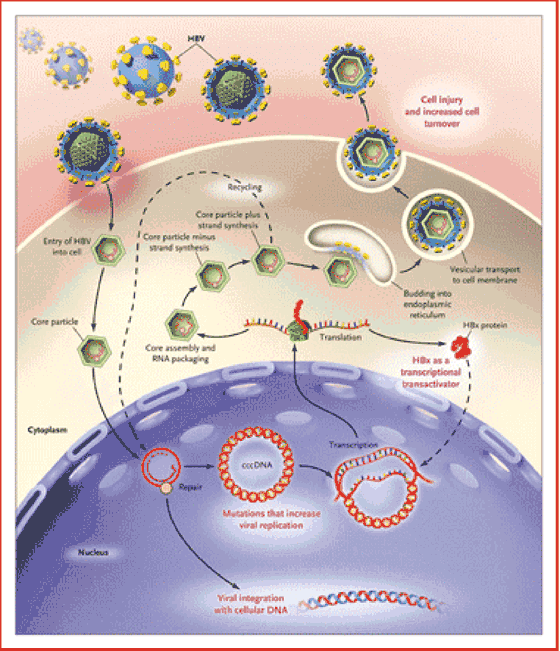
Figure 1. The Life Cycle of HBV and the Molecular and Cellular Mechanisms Associated with the Development of HBV-Related Hepatocellular Carcinoma.
HBV enters the hepatocyte, and the envelope is subsequently removed. Within the nucleus, the partially double-stranded DNA is repaired to form a cccDNA, which serves as the stable template for the transcription of the viral mRNA necessary for productive viral replication. This cccDNA template remains in the nucleus during chronic viral infection and may persist in the liver for the lifetime of the patient.4 Chronic liver injury and increased cell turnover confer a predisposition to hepatocyte transformation, HBV DNA is integrated into the DNA of the hepatocyte, the HBx protein is expressed as a transcriptional transactivator, and mutations in the core promoter region increase viral replication and thus the risk of hepatocellular carcinoma.
Unraveling the life cycle of the virus has been the key to understanding how persistent infection occurs. For example, HBV enters the hepatocyte by means of one or more undefined cell-surface receptors, and the envelope is subsequently removed. The partially double-stranded DNA is then repaired to form a covalently closed circular DNA (cccDNA), which translocates to the nucleus and serves as the stable template for the transcription of the pregenomic messenger RNA (mRNA) and other mRNA species necessary for productive viral replication. This cccDNA template remains in the nucleus during chronic viral infection and may persist in the liver for the lifetime of the patient.4
Antiviral agents such as lamivudine, adefovir dipivoxil, and entecavir work directly on HBV by inhibiting the viral polymerase and thereby suppressing replication. However, the cccDNA template is generally not removed from the nucleus by antiviral therapy, and viral infection persists, since the rate of turnover of infected hepatocytes is quite low. Therefore, cessation of treatment or the development of breakthrough mutations is associated with the prompt resumption of robust viral replication.
The HBV polymerase is highly prone to error and lacks proofreading activity, as do other retroviral reverse transcriptases such as the human immunodeficiency virus. Thus, viral quasispecies will develop during persistent HBV infection, owing to an estimated frequency of 105 to 106 mutations per nucleotide per year.7 In this setting, the use of nucleoside chain terminators such as lamivudine, adefovir dipivoxil, and entecavir runs the risk of selecting for resistant viral strains by suppressing the replication of the major wild-type population. These agents allow noncompetitive drug-resistant quasispecies to replicate and become the dominant viral population. The virologic breakthrough is often associated with a rise in aminotransferase levels, signaling the emergence of drug-resistant strains. The clinical course can be severe, resembling acute hepatitis, particularly in hepatitis B e antigen--negative patients.8
Lamivudine clearly suppresses HBV replication and improves liver histology in patients with chronic hepatitis.9 However, what are the effects of lamivudine in patients with preexisting severe liver disease? In this issue of the Journal, Liaw et al. present the results of a well-designed study that compared the clinical and virologic course of lamivudine treatment with that of placebo in a large group of patients with advanced liver disease or cirrhosis.10 The study was stopped early because of a beneficial effect on disease progression as defined by reduced rates of hepatic decompensation, bleeding esophageal varices, and hepatocellular carcinoma. However, there was an unexplained higher rate of death due to hepatocellular carcinoma in the lamivudine group than in the placebo group during the 2.5-year study period. In addition, YMDD mutations in the polymerase gene developed in 49 percent of the patients in the lamivudine group, and these patients were more likely to have an increase in the Child--Pugh score (an assessment of the severity of liver disease) than were the other patients in the lamivudine group.
Higher death rate from study: "There were 12 deaths among patients originally assigned to receive lamivudine and 4 among those originally assigned to the placebo group. Nine patients died while they were receiving lamivudine (seven during open-label treatment with lamivudine), and seven died during follow-up after treatment. Two patients in the lamivudine group died during double-blind therapy (1 died from preexisting lymphoma; the other drowned after a myocardial infarction), and 14 died after a clinical end point had been reached. These 14 deaths were attributed to hepatocellular carcinoma (8 patients) and an increased Child--Pugh score (6 patients). Eight of the 10 patients originally assigned to receive lamivudine who died after reaching a clinical end point had evidence of YMDD mutations".
The biology of drug-induced HBV mutants may vary in different clinical settings. We know little about the phenotype of the mutants, such as their capacity to replicate, their potential to secrete, and their ability to produce acute and chronic liver injury. Therefore, the use of monotherapy to generate viral strains that have an aggressive liver-injury phenotype merits close monitoring. More important, the emergence of resistant strains may be a function of the ability to inhibit HBV replication in the liver, since drug resistance seems to be less of a problem with an agent, such as adefovir dipivoxil, that is more potent than lamivudine.11 Fortunately, cross-resistance of HBV has not yet become a clinical problem. It is noteworthy that entecavir offers almost complete and sustained inhibition of the replication of woodchuck HBV and greatly reduces the risk of hepatocellular carcinoma and improves survival in the woodchuck model.12 However, entecavir has not yet been approved for clinical use.
The currently available nucleoside analogues (lamivudine and adefovir dipivoxil) are more efficient in promoting the initial decline in the viral load than in altering the second phase of viral elimination, which may require the decay of HBV cccDNA, the eradication of infected hepatocytes, or both. Nevertheless, as shown in the study by Liaw et al., the use of potent antiviral agents such as lamivudine represents a major advance in the treatment of chronic HBV infection and slows the progression of severe liver disease to cirrhosis and hepatocellular carcinoma. Furthermore, there is little doubt that this class of antiviral agents is relatively safe, and prolonged treatment with these agents probably will reduce the incidence of hepatocellular carcinoma. The downside is the long duration of treatment (years or decades), the potential development of clinically severe drug-resistant viral strains with monotherapy, and the very high cost of therapy. The single most important measure for preventing the development of hepatocellular carcinoma is widespread vaccination of susceptible people, since a reduction in the number of chronic carriers of HBV has been associated with a dramatic decrease in the incidence of this devastating disease.13
References
1. El-Serag HB, Davila JA, Petersen JN, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 2003;139:817-823. [Erratum, Ann Intern Med 2004;140:151.]
2. Tagger A, Donato F, Ribero ML, et al. Case-control study on hepatitis C virus (HCV) as a risk factor for hepatocellular carcinoma: the role of HCV genotypes and the synergism with hepatitis B virus and alcohol. Int J Cancer 1999;81:695-699.
3. Moradpour D, Wands JR. Molecular pathogenesis of hepatocellular carcinoma. In: Zakim D, Boyer TD, eds. Hepatology: a textbook of liver disease. 4th ed. Vol. 2. Philadelphia: W.B. Saunders, 2003:1333--54.
4. Ganem D, Prince AM. Hepatitis B virus infection -- natural history and clinical consequences. N Engl J Med 2004;350:1118-1129.
5. Pollicino T, Squadrito G, Cerenzia G, et al. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004;126:102-110.
6. Liang TJ, Baruch Y, Ben-Porath E, et al. Hepatitis B virus infection in patients with idiopathic liver disease. Hepatology 1991;13:1044-1051.[
7. Stuyver LJ, Locarnini SA, Lok A, et al. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology 2001;33:751-757.
8. Hadziyannis SJ, Papatheodoridis GV, Dimou E, Laras A, Papaioannou C. Efficacy of long-term lamivudine monotherapy in patients with hepatitis B e antigen-negative chronic hepatitis B. Hepatology 2000;32:847-851
9. Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med 1999;341:1256-1263.
10. Liaw Y-F, Sung JJY, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med 2004;351:1521-1531.
11. Westland CE, Yang H, Delaney WE IV, et al. Week 48 resistance surveillance in two phase 3 clinical studies of adefovir dipivoxil for chronic hepatitis B. Hepatology 2003;38:96-103.
12. Colonno RJ, Genovesi EV, Medina I, et al. Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection. J Infect Dis 2001;184:1236-1245.
13. Chang M-H, Chen C-J, Lai M-S, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. N Engl J Med 1997;336:1855-1859.
|
|
| |
| |
|
|
|