| |
BILN 2061 HCV Protease Inhibitor Development Held Due to Toxicity
|
| |
| |
"Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients"
Gastroenterology November 2004 • Volume 127 • Number 5
Holger Hinrichsen (I. Medizinische Universitätsklinik, Kiel, Germany) and colleagues
AUTHORS REPORT:
--BILN 2061 was studied for 2 days
--viral RNA reductions of 2--3 log10 copies/mL were achieved in a substantial proportion of patients
--No serious adverse events occurred in all 51 patients. Liver-related laboratory data were not changed.
--In toxicology studies in animals receiving high doses of BILN 2061 for 4 weeks, cardiac toxicity was identified. "As a consequence, further trials in humans are pending until further long-term animal studies have established a safe dose for long-term use".
NATAP reported last year that development of BILN-2061 was discontinued due to animal toxicity. Here is an official publication finally reporting this. The manufacturer appears committed to HCV, howver, and has other HCV drugs in early development, with clinical studies I believe to begin in 2005. In the meantime several HCV drugs are in early study in patients or about to begin study in patients. NM283 is an HCV polymerase inhibitor showing good antiviral efficacy as monotherapy and in recently presented study at AASLD in October in combination with peginterferon. Vertex's HCV protease inhibitor is just starting its first study in patients at the end of 2004. Schering Plough has a protease inhibitor although I haven't heard its development status recently. Additional HCV drugs are in early development while much pharmaceutical R&D are committed to HCV. But it will be several years at a minimum before we might see the first new HCV drug & guaranteed that peginterferon will have to be used with any new drugs for the foreseeable future.
ABSTRACT
Background & Aims: Novel, potent, and well-tolerated hepatitis C virus (HCV) drugs are needed. BILN 2061 is a potent and specific inhibitor of HCV serine protease in vitro. Preclinical toxicology data and studies in healthy volunteers supported the administration of BILN 2061 to patients with HCV infection.
Methods: The antiviral efficacy, pharmacokinetics, and tolerability of 25, 200, and 500 mg BILN 2061 twice daily given as monotherapy for 2 days in 31 patients infected with chronic genotype 1 HCV infection and with minimal liver fibrosis (Ishak score of 0--2) were assessed in a placebo-controlled, double-blind pilot study. In 2 subsequent placebo-controlled studies of similar design, 200 mg BILN 2061 twice daily was administered for 2 days to 10 patients with advanced liver fibrosis (Ishak score of 3 or 4) and to 10 patients with compensated cirrhosis (Ishak score of 5 or 6).
Results: Viral RNA reductions of 2--3 log10 copies/mL were achieved in most of the patients. There was a trend toward a higher number of patients receiving 500 mg BILN 2061 achieving a viral RNA reduction >=3 log10 copies/mL as compared with patients receiving 25 mg BILN 2061. Advanced fibrosis or compensated cirrhosis did not affect the antiviral efficacy of BILN 2061. BILN 2061 was well tolerated in all studies.
Conclusions: BILN 2061 is a well-tolerated and very active compound that reduced serum viral RNA concentrations after 2 days of treatment in patients infected with genotype 1 HCV independent of the degree of fibrosis. Nevertheless, further clinical trials are on hold pending resolution of animal toxicity issues.
AUTHOR DISCUSSION
The results of these 3 exploratory studies indicate that BILN 2061, administered twice daily over a 2-day period, is effective and well tolerated in patients infected with genotype 1 HCV. It should be noted that the baseline patient characteristics were typical for HCV-infected patients except for the underrepresentation of women, which was due to the reproduction-related exclusion criteria. The efficacy and tolerability of BILN 2061 were not limited by the stage of fibrosis, even if cirrhosis had developed.
Transient viral RNA reductions of 2--3 log10 copies/mL were achieved in a substantial proportion of patients. A dose-response relationship was detectable over the dose range investigated; while 200- and 500-mg doses of BILN 2061 appeared to be equally effective, not all patients in the 25-mg dose group experienced a viral RNA reduction of >=2 log10 copies/mL. Virologic responses were similar in interferon alfa nonresponder and treatment-naive patients as well as in patients with high and low baseline viral RNA concentrations. Patients infected with genotype 1a or 1b HCV appeared to respond equally well to BILN 2061. Although the NS3 serine protease is within a highly conserved region, first clinical results indicate that the antiviral potency of this compound is less in patients infected with genotype 2 and 3 HCV, indicating that other factors might interact with the binding of BILN 2061 to the NS3 serine protease.
The results of the transcription-mediated amplification assay and the rapid viral rebound, which occurred after therapy was completed, suggest that low-level HCV replication continued during the 2 days of BILN 2061 administration. It is not known whether HCV can persist as a latent virus and, if so, which cells or tissue sites it is harbored in. Because it is unlikely that 2 days of therapy, albeit with a very potent drug, would eradicate the virus, these findings are not unexpected. Further studies would be needed to determine if prolonged therapy with BILN 2061, either as monotherapy or in combination with other anti-HCV drugs, leads to a longer-lasting effect. No BILN 2061-resistant strains emerged in these short-term studies, but there are initial reports that BILN 2061-resistant viral strains emerge in the replicon assay.31 Further studies would also show whether resistant strains emerge in humans.
The major initial effect of interferon alfa is to block HCV production or the release of virions from infected cells.32 The level of inhibition achieved with a single dose of interferon alfa of 10 million IU/day is 95% and for a dose of 15 million IU/day is 96%. The average estimated virion half-life is 2.7 hours, and the pretreatment production and clearance rate is 1012 virions/day. The 2-day period of this study was chosen so that BILN 2061 was administered for the duration of approximately 10 HCV replication cycles. The HCV RNA concentration decreases to a level of 2000 copies/mL after 3 weeks in patients who respond rapidly to interferon alfa. By contrast, after 1 day, the viral RNA concentration was reduced to below the level of detection of the Amplicor assay (1500 copies/mL) in the majority of patients treated with 200 mg BILN 2061. The lag time before viral RNA reductions were observed was only a few hours, if at all, in this study, as compared with 8--9 hours with interferon alfa. The observation that similar viral RNA reductions were achieved in treatment-naive patients and those who had failed previous therapy with interferon alfa-containing regimens supports that BILN 2061 has a different mechanism of action against HCV.
The AST and ALT levels did not change during these trials. This is not unexpected, because viral RNA concentrations rebounded in all cases after the 2-day administration of BILN 2061 had been completed. In addition, it is likely that the duration of viral reduction was too short to reduce hepatic inflammation.
No serious adverse events occurred in all 51 patients. Liver-related laboratory data were not changed. A broad range of side effects occurred, and further clinical studies will be necessary to carefully characterize the side effect profile of this compound. The provision of BILN 2061 in polyethylene glycol may have accounted for some gastrointestinal symptoms. In recent toxicology studies in animals receiving high doses of BILN 2061 for 4 weeks, cardiac toxicity was identified. As a consequence, further trials in humans are pending until further long-term animal studies have established a safe dose for long-term use.
In conclusion, orally administered BILN 2061 is highly active against HCV genotype 1 infections in patients with all degrees of liver fibrosis, including cirrhosis.
INTRODUCTION
Infection with hepatitis C virus (HCV) is a common cause of liver disease, affecting between 1% and 3% of the world's population. Liver failure due to HCV is a leading cause of liver transplantation in the United States1 and Europe. Genotype 1 HCV is the most common genotype in the United States, Europe, and Japan; it accounted for 74% of infections in the National Health and Nutrition Examination Survey in the United States, 69% of cases in a European survey, and more than 77% in special risk groups such as patients on hemodialysis.
Treatment options for HCV infection have improved over recent years but are still less than perfect, especially for patients infected with genotype 1 HCV. Toxicities associated with interferon alfa therapy include a flu-like syndrome, neutropenia, thrombocytopenia, and depression. Hemolytic anemia is the main adverse effect associated with ribavirin. In genotype 2 and 3 HCV infections, a sustained virologic response is achieved in approximately 80% of patients treated with pegylated interferon and ribavirin; in genotype 1 HCV infections, the sustained virologic response is <50%. Dose reduction or discontinuation of therapy were relatively common in both the European and US meta-analyses of studies of interferon alfa with or without ribavirin. As a result of these limitations, a considerable proportion of patients do not derive long-term benefit from these therapeutic regimens.
Hence, there is a considerable need for novel, potent, and well-tolerated anti-HCV drugs. The progression of fibrosis and established cirrhosis provide an indication of the need for, and urgency of, therapy.
HCV is a small, enveloped virus that contains a single-stranded RNA genome; the RNA encodes a polyprotein of at least 3000 amino acids. The polyprotein is a precursor of several structural and nonstructural proteins. The structural proteins are released via proteolysis by host signal peptidases, whereas the 2 viral proteases, NS2 and NS3/NS4a, are responsible for the nonstructural polyprotein processing. The NS3/NS4a serine protease mediates proteolysis at the NS3/NS4a, NS4a/NS4b, NS4b/NS5a, and NS5a/NS5b junctions. Thus, the NS3/NS4a serine protease plays a key role in HCV polyprotein processing and, therefore, viral replication. In 1996, the crystal structure of the NS3/NS4a serine protease of HCV was determined by 2 different groups. Because this serine protease is essential for viral replication, identifying inhibitors of this enzyme is a logical method of developing agents for the treatment of HCV infection.
BILN 2061 is a peptidomimetic and a potent and specific inhibitor of HCV serine protease, as described previously.24 Preclinical studies have shown that BILN 2061 is orally bioavailable in several animal species. Preclinical toxicology data supported the administration of repeated doses of BILN 2061 in humans for up to 2 weeks. No evidence of genotoxicity was obtained in vitro. In light of its potent activity in vitro, its good pharmacokinetics in animal models, and its adequate preclinical safety profile, BILN 2061 was selected for in-depth, clinical evaluation in humans as a novel antiviral compound with considerable therapeutic potential for the treatment of HCV infection. BILN 2061 (single oral doses of 5--2400 mg/day) or placebo was administered to healthy male volunteers in a dose-escalation study.25 No drug-related adverse effects were noted at doses up to 2000 mg/day, and BILN 2061 was well tolerated. The drug is rapidly absorbed, and BILN 2061 has a half-life of approximately 4 hours. Because there are no in vivo animal models to assess the potential pharmacologic response to BILN 2061, it was necessary to evaluate its antiviral effects in humans.
Three separate studies of BILN 2061 were performed, all focusing on patients with chronic hepatitis C infected with genotype 1. Study 1 evaluated the antiviral efficacy and safety of different doses of BILN 2061 given for 2 days to patients with mild liver disease and mild or no liver fibrosis (Ishak score of 0--2). Study 2 focused on the 200-mg dose of BILN 2061 given for 2 days to patients with more advanced liver fibrosis (Ishak scores of 3 or 4), and study 3 focused on the same dose regimen given to patients with compensated cirrhosis (Ishak scores of 5 or 6).
All 3 trials were performed in accordance with the principles stated in the Declaration of Helsinki. Ethics committee approval was obtained at each study site, and all patients gave written informed consent.
Study 1 design
Patients were eligible for enrollment if they were women or men aged 18 years or older with chronic genotype 1 HCV infection. The line probe assay was used to determine the genotype of the viral infection. A liver biopsy specimen showing changes consistent with chronic HCV infection had to have been performed within the previous 12 months. At screening, the HCV load had to be >50,000 copies/mL serum (Amplicor assay; Roche, Penzberg, Germany). Women were excluded if they were breast-feeding or at risk of pregnancy; men had to use an adequate form of contraception if their partner was of childbearing potential. Patients were ineligible for the study if there was evidence of bridging fibrosis or higher-grade fibrosis (fibrosis with an Ishak score >=3 or Metavir score >=2). They were not enrolled if there were other or additional reasons for chronic liver disease, including the presence of other hepatitis-causing viruses and/or a history of alcohol abuse within the previous 12 months and/or evidence of Child's B or C liver disease at screening. No other antiviral or antimicrobial or investigational therapies were allowed during the study (screening, pretreatment, and treatment phases).
Patients were excluded if, at screening, their baseline alanine aminotransferase (ALT)/aspartate aminotransferase (AST) plasma levels exceeded the upper limit of normal by more than 5-fold (>5 times the upper limit of normal) or their total bilirubin or alkaline phosphatase levels were 1.5 times the upper limit of normal. Other exclusion criteria included coinfection with human immunodeficiency virus, a platelet count <100,000/mm3, a white blood cell count <2000 cells/mm3, any clinically significant laboratory abnormalities, and a positive test result for illicit or nonprescription drugs.
Patients were treated as inpatients from the day before the start of treatment to the day after the last dose of the study drug and followed thereafter as outpatients until the end of the study (days 10--14).
Twice-daily doses of 200, 500, and then 25 mg BILN 2061 or placebo were administered to 10 patients per dose for 2 days; the randomization was 4:1. Placebo was given in a double-blind manner. The starting dose of 200 mg was selected based on replicon in vitro data in comparison with the effective human dose of interferon alfa.22 The 500-mg dose was chosen to evaluate the upper limit of safety established by the phase 1 data. After data using the first 2 doses were obtained, the 25-mg dose was studied to determine the lower limit of efficacy of BILN 2061. For solubility reasons, 3 mL of the drug was administered in a polyethylene glycol 400/ethanol solution. The amount of ethanol administered was 1.2 mL/day. All doses of BILN 2061 and placebo were given as directly observed therapy. There was a 10-day follow-up period.
Blood was drawn for HCV RNA determination and pharmacokinetic analysis at short intervals on both treatment days (0, .5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, and 14 hours), on days 3 and 4, and between days 5--7 and days 10--14.
Safety monitoring was conducted from the time of screening to the end of the follow-up period (10 ± 2 days). A safety interview was performed at short intervals on both treatment days (0, 1, 2, 4, 8, and 14 hours), on days 3 and 4, and between days 5--7 and days 10--14. An electrocardiogram was obtained on day -1, on day 3, and at the end of the study (days 10--14). Routine laboratory parameters, including ALT and AST levels, were determined during screening; at days 1, 2, and 3; and at the end of the study (days 10--14).
Study 2 design
Ten patients with genotype 1 HCV infection and advanced liver fibrosis were enrolled in study 2. Patients had to have an Ishak score of 3 or 4 or a Metavir score of 2 or 3 (except a Metavir score of 3 and an Ishak score of 5), based on the results of a biopsy performed within 12 months before starting the study. Inclusion and exclusion criteria differed from those of study 1 regarding liver histology and the range of AST, ALT, and bilirubin levels. Patients were allowed to participate if their ALT and AST levels did not exceed 10 times the upper limit of normal or if their bilirubin levels were <2 mg/dL.
Study 3 design
Ten patients with genotype 1 HCV infection and compensated liver cirrhosis were studied in study 3. Patients had to have an Ishak score of 5 or 6 or a Metavir score of 3 or 4 (except a Metavir score of 3 and an Ishak score of 4), based on the results of a biopsy performed within 36 months before starting the study. Inclusion and exclusion criteria differed from those of study 2 regarding liver histology and platelet count. Only patients with a platelet count >80,000/mm3 were included. Patients had to have grade A liver cirrhosis according to the Child-Turcotte-Pugh classification and were excluded if there was prior evidence of decompensated liver cirrhosis. Ultrasonography was performed at screening to exclude patients with hepatocellular carcinoma.
In both studies 2 and 3, the randomization ratio was 4:1.
Laboratory determinations
HCV viral RNA concentrations were measured with Cobas Amplicor HCV Monitor version 2.0. In addition, branched DNA (bDNA) version 3.0 (Bayer, Tarrytown, NY) and transcription-mediated amplification (Bayer) assays were used in all 3 trials. The lower limit of quantification for the Amplicor assay was 1500 RNA copies/mL or 600 IU/mL and for the bDNA assay was 3200 RNA copies/mL or 615 IU/mL. The limit of detection for the transcription-mediated amplification assay was 50 RNA copies/mL or 5 IU/mL.26,27
Plasma samples for pharmacokinetics, including peak plasma concentration, peak time, and area under the curve, were analyzed by liquid/mass spectrometry (unpublished data, Darnow J et al. BILN 2061 ZW: a method for the quantitation of BILN 2061 ZW in human plasma by HPLC-MS/MS. Validation and stability data. Boehringer Ingelheim, 2002).
Patient demographics
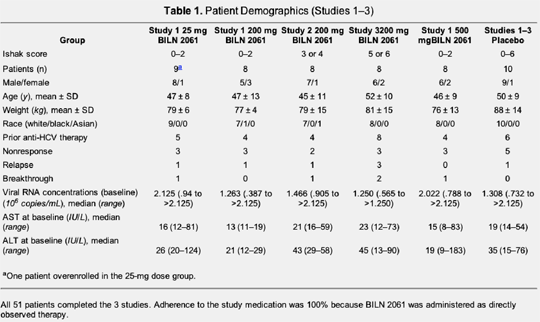
Thirty-one patients were enrolled in study 1, 10 in study 2, and 10 in study 3. The clinical features and demographics of the 51 patients in the 3 studies are shown in Table 1. The majority of patients were men (80%), and most were white (96%). The average age was 48 years but was slightly higher among patients with cirrhosis (study 3; 52 years). Mean body weight was 80 kg. In all 3 studies, more than one half of the patients (31 patients; 61%) had received antiviral therapy with interferon alfa with or without ribavirin in the past but had either failed to respond (n = 19) or experienced viral breakthrough (n = 5) or relapse (n = 7). Mean pretreatment viral RNA concentration was 1.486 × 106copies/mL (ranging from .338 × 106 copies/mL to >2.125 × 106 copies/mL) and was similar in the different patient groups. The mean AST and ALT values were 22.5 IU/mL and 39.8 IU/mL, respectively.
|
|
| |
| |
 |
|
| |
| |
Virologic efficacy
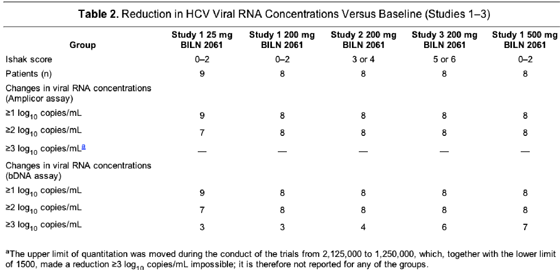
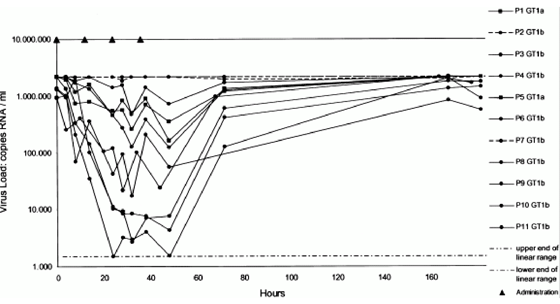
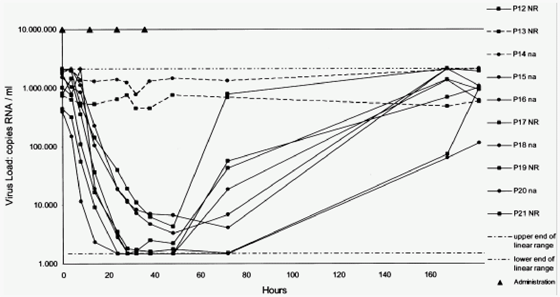
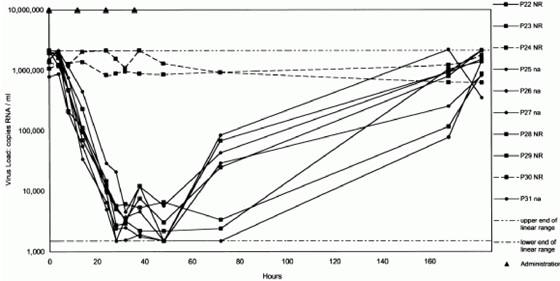
In the 25-mg dose group, all 9 patients achieved a viral RNA reduction of >=1 log10 copies/mL, 7 (78%) had a reduction of >=2 log10 copies/mL, and 3 (33%) achieved, by the bDNA assay, a reduction of >=3 log10 copies/mL (Table 2 and Figure 1). These 3 patients had the 3 highest blood plasma levels seen in this dose group (see following text). The HCV load decreased by >=2 log10 copies/mL, as measured by the Amplicor assay and the bDNA assay, in all patients who were treated with BILN 2061 (200 or 500 mg) (Table 2 and Figures 2 and 3). Thirteen of 24 patients (54%) in the 200-mg dose group and 7 of 8 (88%) in the 500-mg dose group had a reduction of >=3 log10 copies/mL by the bDNA assay. No changes were observed in the viral RNA concentrations of the 10 control patients. No difference in viral responses was seen between patients previously treated with interferon alfa-containing regimens (nonresponders) or treatment-naive patients (Figures 2 and 3) as well as between patients who had high or low viral RNA concentrations at baseline. In addition, genotype 1a and 1b strains responded equally to administration of BILN 2061 (Figure 1).
|
|
| |
| |
 |
|
| |
| |
Figure 1. Virologic efficacy of 25 mg BILN 2061 twice daily (Amplicor assay) in patients with minimal liver fibrosis: study 1. Dotted lines represent control patients, solid lines represent treated patients, upper and lower intermittent lines represent the upper and lower limits of detection of the Amplicor assay, black triangles represent administration of BILN 2061, squares represent genotype 1a, and circles represent genotype 1b.
|
|
| |
| |
 |
|
| |
| |
Figure 2. Virologic efficacy of 200 mg BILN 2061 twice daily (Amplicor assay) in pretreated and drug-naive patients with minimal liver fibrosis: study 1. Dotted lines represent control patients, solid lines represent treated patients, circles represent drug-naive patients (na), squares represent patients who did not respond to anti-HCV therapy (NR), upper and lower intermittent lines represent the upper and lower limits of detection of the Amplicor assay, and black triangles represent administration of BILN 2061.
|
|
| |
| |
 |
|
| |
| |
Figure 3. Virologic efficacy of 500 mg BILN 2061 twice daily (Amplicor assay) in patients with minimal liver fibrosis: study 1. Dotted lines represent control patients, solid lines represent treated patients, circles represent drug-naive patients (na), squares represent patients who did not respond to anti-HCV therapy (NR), upper and lower intermittent lines represent the upper and lower limits of detection of the Amplicor assay, and black triangles represent administration of BILN 2061.
|
|
| |
| |
 |
|
| |
| |
At all time points, the transcription-mediated amplification assay was positive in all patients. The HCV viral RNA concentrations returned to baseline within 1--7 days of completing 2 days of treatment with BILN 2061. Substantial changes in the secondary efficacy parameters, AST and ALT levels, were not observed.
Pharmacokinetics
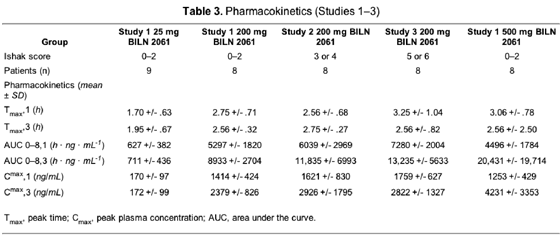
The range of the assay for determining the level of BILN 2061 was .25--100 ng/mL. The pharmacokinetics following the first and third doses of the compound are presented in Table 3. The majority of peak time values were obtained between 2 and 4 hours following administration of the morning dose of BILN 2061 on both days 1 and 2.
|
|
| |
| |
 |
|
| |
| |
For the 25-mg dose group, peak plasma concentration and area under the curve levels were only slightly increased on day 2 compared with day 1. However, these parameters were, on average, more than 70% higher than on day 1 for the 200-mg dose group and increased between 3- and 4-fold, on average, for the 500-mg dose group on day 2.
Analysis of data from the lowest dose group (25 mg twice daily) showed that the 3 best responders in this group, who all experienced a viral RNA reduction of >=3 log10 copies/mL, had peak plasma concentration levels >150 ng/mL and area under the curve levels for the first 8 hours of >500 h · ng · mL-1. By contrast, all other patients in this group had lower plasma levels of BILN 2061. This relationship was not observed in the other 2 dose groups. Interpatient variability was higher in patients with advanced liver fibrosis (study 2) or cirrhosis (study 3).
Safety
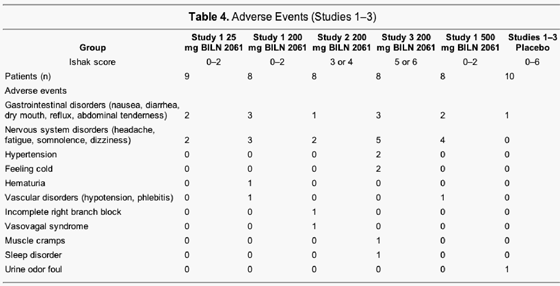
BILN 2061 was well tolerated. No relevant drug-induced changes in vital signs, routine laboratory findings, or electrocardiograms were observed during the 3 studies. In particular, there was no indication of BILN 2061-related liver injury, and increases in AST or ALT levels or changes in other liver-related laboratory data (serum albumin levels, prothrombin time, and bilirubin levels) were not observed in patients treated with BILN 2061. No serious adverse events occurred during the study or the follow-up period. The rate of total adverse events during the study was slightly higher in the 200- and 500-mg dose groups than in the 25-mg dose and placebo groups. Possible drug-related adverse events included mild constitutional symptoms, such as headache, fatigue, and somnolence, and mild gastrointestinal symptoms, such as diarrhea and nausea (Table 4). All adverse events had resolved by the end of the follow-up period. Urine pH decreased in one third of treated and control patients, possibly due to the polyethylene glycol/ethanol solution of the compound.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
|
|
|