| |
HBV/HIV Coinfection & Tenofovir
|
| |
| |
"Efficacy of Tenofovir Disoproxil Fumarate in Antiretroviral Therapy--Naive and --Experienced Patients Coinfected with HIV-1 and Hepatitis B Virus"
Gregory J. Dore,1 David A. Cooper,1 Anton L. Pozniak,2 Edwin DeJesus,3 Lijie Zhong,4 Michael D. Miller,4 Biao Lu,4 and Andrew K. Cheng,4 for the 903 and 907 Study Teams
1National Centre in HIV Epidemiology and Clinical Research, University of New South Wales, Sydney, Australia; 2Chelsea and Westminster Hospital, London, United Kingdom; 3IDC Research Initiative, Altamonte Springs, Florida; 4Gilead Sciences, Foster City, California
ABSTRACT
Coinfection with human immunodeficiency virus type 1 (HIV-1) increases the risk of hepatitis B virus (HBV)--associated progressive liver disease. Lamivudine has potent activity against both HIV-1 and HBV; however, lamivudine-resistance mutations in HBV frequently develop.
Substudies of the safety and efficacy of tenofovir disoproxil fumarate (tenofovir DF) for patients coinfected with HIV and HBV were undertaken within 2 phase 3 randomized controlled trials involving antiretroviral therapy--experienced (study 907) and --naive (study 903) HIV-infected populations. Inclusion criteria were detection of hepatitis B surface antigen, an HBV DNA level >106 copies/mL at baseline, and HBV DNA specimens available at week 24 (study 907) and week 48 (study 903).
In study 907, the mean decrease in HBV DNA was 4.9 log10, after 24 weeks, for 10 patients randomized to receive tenofovir DF, compared with a mean increase of 1.2 log10 for 2 patients randomized to receive placebo (P = .041). The mean decrease in HBV DNA during tenofovir DF treatment was similar for patients with wild-type (5.3 log10) and lamivudine-resistant (4.6 log10) HBV strains. In study 903, the mean decrease in HBV DNA was 3.0 log10, after 48 weeks, for 6 patients randomized to receive lamivudine, compared with 4.7 log10 for 5 patients randomized to receive lamivudine and tenofovir DF (P = .055). Four patients developed tyrosine-methionine-aspartate-aspartate mutations, all in the lamivudine-only treatment arm. (note from Jules Levin: combination therapy for HBV has not yet been well studied and appearances of no additional efficacy of 2 drugs compared to 1 drug may not be accurate since combination therapy has not yet been well studied and due to limitations in HBV viral load assays used. There are suggestions from existing data that combination therapy may have benefits but it may depend on the drugs used and additional considerations).
Tenofovir DF has potent anti-HBV efficacy in antiretroviral therapy--experienced and --naive individuals coinfected with HIV and HBV.
INTRODUCTION
Improved survival resulting from the success of highly active antiretroviral therapy (HAART) has enabled chronic viral hepatitis to become a major source of comorbidity in HIV-infected populations. HIV modifies the natural history of hepatitis B virus (HBV), with higher rates of chronic HBV infection, replicative disease, and progression to advanced liver disease. A higher HBV load and reactivation of HBV infection in chronic carriers who have lost detectable serum levels of hepatitis B e antigen (HBeAg) are also well described in the HIV-HBVcoinfected population.
Although HBV does not appear to influence progression of HIV disease or response to HAART, the incidence of antiretroviral therapyassociated hepatotoxicity is increased 3-fold in individuals with underlying chronic HBV infection. Thus, strategies to limit both HBV-associated progressive liver disease and hepatotoxicity are required for the management of HIV-HBV coinfection.
Lamivudine has potent activity against both HIV-1 and HBV. Among people with HIV-HBV coinfection, HBV DNA levels decrease by 23 log10 copies/mL; in 40%-80% of such people, the HBV DNA level becomes undetectable (<400 copies/mL), as determined by polymerase chain reaction (PCR) analysis, during the initial 12 months of lamivudine therapy. However, lamivudine resistance, which occurs through the development of HBV DNA polymerase tyrosine-methionine-aspartate-aspartate (YMDD) motif mutations, is common in both HBV-monoinfected and HIV-HBVcoinfected populations, reaching a level of 90% resistance among HIV-HBVcoinfected individuals after 4 years of therapy. Furthermore, hepatitis "flares" and fulminant hepatic failure have been described in association with lamivudine resistance and withdrawal in individuals with HIV-HBV coinfection.
Tenofovir disoproxil fumarate (tenofovir DF) is an oral prodrug of tenofovir, a novel, acyclic nucleotide analogue with in vitro activity against HIV-1, HIV-2, and HBV. Activity against HBV in vitro has been demonstrated for both wild-type (wt) HBV and lamivudine-resistant HBV. Several studies have reported the efficacy of antiviral therapy with tenofovir DF among antiretroviral therapyexperienced HIV-HBVcoinfected individuals. However, these studies have involved small study populations receiving open-label therapy.
Two phase 3 randomized, double-blind, placebo-controlled trials recently examined the safety and efficacy of tenofovir DF among antiretroviral therapy experienced (study 907) and naive (study 903) HIV-1 infected patients. Substudies of study 907 and study 903 were undertaken to examine the safety and efficacy of tenofovir DF among antiretroviral therapy experienced and naive HIV-HBVcoinfected individuals. Individuals in study 907 were randomized to receive tenofovir DF or placebo, and individuals in study 903 were randomized to receive antiretroviral therapy regimens that included lamivudine plus tenofovir DF versus lamivudine alone as agents active against HBV.
Study population and design of study 907. Recruitment of patients for the study began in October 1999 and continued until June 2000, at 75 HIV clinics in Western Europe, North America, and Australia [30]. Informed consent was obtained from the patients, and human experimentation guidelines of the US Department of Health and Human Services were followed in the conduct of study 907. Patients (age range, 1865 years) were eligible for inclusion in the study if they had received antiretroviral therapy (4 agents) for 8 weeks before randomization and if they had stable plasma levels of HIV-1 RNA (40010,000 copies/mL), as determined using the Roche Amplicor HIV-1 Monitor UltraSensitive test (version 1.0; lower limit of quantification, 50 copies/mL). There were no CD4 cell count restrictions for entry into the study. Chronic liver disease was not a criterion for exclusion; however, a platelet count of <50,000 platelets/mm3, a total bilirubin concentration >1.5 mg/dL, an alanine aminotransferase (ALT) level 108 U/L, and an aspartate aminotransferase level 90 U/L were criteria for exclusion. Individuals who had previously received tenofovir DF or adefovir dipivoxil were also excluded from the study.
A total of 552 eligible participants were stratified according to HIV-1 RNA level at baseline (<5000 copies/mL or 5000 copies/mL), CD4 cell count at baseline (<200 cells/mm3 or 200 cells/mm3), and number of antiretroviral drugs received before entry into the study (<4 drugs or 4 drugs). The patients then were randomized (2 : 1) to receive either tenofovir DF (300 mg) or placebo, in addition to their existing background antiretroviral regimen. Changes to the background regimen were discouraged during the first 24 weeks of the study. After week 24, all participants received open-label tenofovir DF for the remainder of the 48-week study. The incidence of grade 3 or 4 clinical adverse events or laboratory abnormalities was evaluated as safety end points at baseline and at weeks 2, 4, 8, 12, 16, 20, 24, 32, 40, and 48.
Study population and design of study 903. Recruitment of patients for the study began in April 2000 and continued until January 2001, at 81 centers in Latin America, Europe, and the United States. Informed consent was obtained from the patients, and human experimentation guidelines of the US Department of Health and Human Services were followed in the conduct of study 903. Adult patients who were eligible for inclusion in the study had plasma levels of HIV-1 RNA >5,000 copies/mL, were treatment naive, and had stable hematologic and renal parameters. There was no minimum CD4 cell count criterion for entry into the study. Chronic liver disease was not a criterion for exclusion from the study; however, a platelet count of <50,000 platelets/mm3, a total bilirubin concentration >1.5 mg/dL, an ALT level 108 U/L, and an aspartate aminotransferase level 90 U/L were exclusion criteria.
Six hundred patients were randomly assigned, in a 1 : 1 ratio, to receive either tenofovir DF (300 mg once daily) or stavudine (40 mg twice daily, or 30 mg twice daily if body weight was <60 kg; Bristol-Myers Squibb) plus corresponding placebo, in combination with lamivudine (150 mg twice daily; GlaxoSmithKline) and efavirenz (600 mg every day; Bristol-Myers Squibb). Nevirapine (200 mg twice daily; Boehringer Ingelheim) could be substituted for efavirenz in the event of intolerable efavirenz-associated neuropsychiatric toxicity. Patients were stratified according to the HIV-1 RNA level (>100,000 copies/mL or 100,000 copies/mL) and CD4 cell count (<200 cells/mm3 or 200 cells/mm3) determined during screening. Clinical and laboratory evaluations were performed at screening, before baseline, at baseline, at weeks 2 and 4, and then every 4 weeks through week 48.
Substudy methodologies. For substudies of both study 907 and 903, the following methodologies were undertaken. Nearly all participants who were enrolled in the substudies had baseline serum specimens prospectively tested for HBV surface antigen (HBsAg; Abbott Auszyme Monoclonal EIA or Abbott AxSYM). Detection of HBV DNA (Roche Amplicor HBV Monitor Test), HBeAg (Diasorin ETI-EBK Kit), and hepatitis B e antibody (Diasorin ETI-EBK Kit) was performed prospectively in serum specimens obtained every 12 weeks through week 48. Genotypic analysis of HBV DNA for the detection of mutations within the YMDD motif of the DNA polymerase was performed for specimens, obtained both at baseline and at week 48, for which the HBV DNA level was >1000 copies/mL. The HBV polymerase reverse-transcriptase domain rt 1 and rt 344 (1032 nucleotides) was sequenced for these analyses.
Criteria for inclusion of patients in the substudies were detection of HBsAg at baseline, an HBV DNA level >106copies/mL at baseline, and HBV DNA specimens available for assessment at week 24 (study 907) and week 48 (study 903). The primary end point for both substudies was the mean change in the HBV DNA level, from the level at baseline, during the initial 24 weeks (study 907) or 48 weeks (study 903) of the study. Secondary end points included the mean change in the ALT level, the proportion of patients with YMDD-resistant strains at week 48, and loss of HBeAg.
BASELINE CHARACTERISTICS
|
|
| |
| |
 |
|
| |
| |
RESULTS
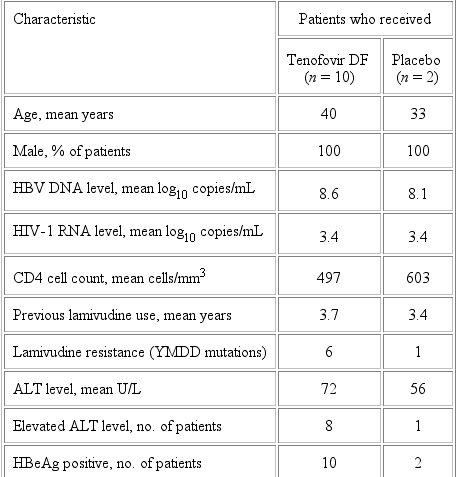
Study 907. Screening for HBsAg was undertaken for 539 (98%) of the 552 patients in study 907, and 23 (4.3%) of these 539 patients were HBsAg positive. Twelve (52%) of the 23 HBsAg-positive patients had an HBV DNA level >106 copies/mL at baseline and had specimens available for analysis at week 24; these patients formed the substudy population. HBeAg was present in all 12 patients, lamivudine-resistant strains were present in 7 of 12 patients, and the mean HBV DNA level was 8.5 ¥ 108 copies/mL.
At week 24, the mean decrease in the HBV DNA level, from baseline, was 4.9 log10 copies/mL for the 10 patients who received tenofovir DF, compared with a mean increase of 1.2 log10 copies/mL for the 2 patients who received placebo (P = .041). For the 10 patients who were originally randomized to receive tenofovir DF, the decrease in the HBV DNA level was maintained (4.5 log10 copies/mL) through week 48. At week 24, the mean decrease in the HBV DNA level of the patients who received tenofovir DF was similar to that noted for patients with wt HBV strains (n = 4; 5.3 log10 copies/mL) and lamivudine-resistant strains (n = 6; 4.6 log10 copies/mL).
Of the 8 patients who had abnormal ALT levels (greater than the upper limit of the range considered to be normal) at baseline and who received tenofovir DF, 2 had ALT levels that normalized during the 48 weeks of the study. One patient who received tenofovir DF underwent HBeAg seroconversion, whereas an additional patient lost HBeAg but remained anti-hepatitis B e (HBe) negative. All YMDD mutations that were present at baseline and were detectable were maintained, and no new conserved mutations were detected in HBV polymerase through week 48.
Among patients who received tenofovir DF, 3 serious adverse effects that were not considered to be related to the study drug occurred (i.e., fever, schizophrenic reaction, and pneumonia), but none of these patients discontinued participation in the study as a result. One patient who received tenofovir DF (in addition to stavudine, delavirdine, and abacavir) developed a grade 3 elevation in the ALT level at week 44 and discontinued receiving study drugs. The CD4 cell count of this patient had increased from 364 cells/mm3 at baseline to 481 cells/mm3 at week 40, but the HBV DNA level had decreased from 70,677,000 copies/mL to 1680 copies/mL. The hepatic flare resolved after 12 weeks and was not associated with hepatic decompensation. A second patient discontinued participation in the study before week 48, after moving to another city.
Mean HIV-1 RNA levels at baseline were similar in the substudy population and the overall study 907 population (3.4 log10 copies/mL). The HIV-1 RNA response during treatment with tenofovir DF was similar through week 24 in the overall study population (-0.6 log10 copies/mL) and the substudy patients who were infected with HIV and HBV (-0.8 log10 copies/mL). The change in the mean CD4 cell count at week 24 was similar for the 2 patients who received placebo (603659 cells/mm3) and the 10 patients who received tenofovir DF (497532 cells/mm3).
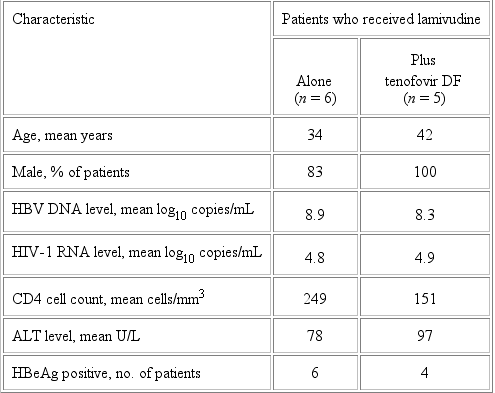
Study 903. Screening for HBsAg was undertaken for 599 of the 600 patients in the study, and 23 (3.8%) of these 599 patients were found to be HBsAg positive. Eleven (48%) of these 23 patients had an HBV DNA level >106 copies/mL at baseline and had specimens from week 48 available for analysis; these patients formed the substudy population. Characteristics, at baseline, for the 6 patients who were randomized to receive stavudine, lamivudine, and efavirenz, and for the 5 patients who were randomized to tenofovir DF, lamivudine, and efavirenz, are shown in table 2. HBeAg was present in 10 of 11 patients, and the mean HBV DNA level was between 108 copies/mL and 109 copies/mL.
|
|
| |
| |
 |
|
| |
| |
At week 48, the mean decrease in the HBV DNA level was 3.0 log10 copies/mL for the 6 patients who received lamivudine, compared with 4.7 log10 copies/mL for the 5 patients who received lamivudine and tenofovir DF (P = .055). Five of 6 patients who received lamivudine alone had detectable HBV DNA (>1000 copies/mL) at week 48, and 4 of these 5 patients developed YMDD mutations (M204V + L180M developed in 3 patients, and M204I developed in 1 patient). For the 4 patients who had YMDD mutations, the mean increase in the HBV DNA level was 2.3 log10 copies/mL, as determined from the HBV DNA nadir. In contrast, 4 of 5 patients who received lamivudine and tenofovir DF had undetectable HBV DNA at week 48, and none of these patients had YMDD mutations develop. Evaluation of specimens obtained at baseline revealed no YMDD mutations in the 11 study patients. Of 4 patients who had abnormal ALT levels at baseline and who received tenofovir DF and lamivudine, 3 had ALT levels that normalized during the 48 weeks of the study. In contrast, of 4 patients who had abnormal ALT levels at baseline and who received lamivudine without tenofovir DF, 1 had ALT levels normalize during the 48 weeks. Three patients (1 from the tenofovir DFlamivudine treatment arm and 2 from the lamivudine treatment arm) had normal ALT levels at baseline.
One serious adverse effect (myelitis) occurred among patients who received lamivudine, and one serious adverse effect (bacterial infection) occurred among patients who received lamivudine and tenofovir DF. Four patients who received lamivudine and 2 patients who received lamivudine and tenofovir DF developed grade 3 or 4 elevations of ALT through week 48. These hepatic flares were not associated with hepatic decompensation.
The mean HIV-1 RNA level at baseline was similar in the substudy population and the overall population of study 903 (4.84.9 log10 copies/mL). The HIV-1 RNA response of patients coinfected with HIV and HBV (3.0 log10 copies/mL) was similar to that of the overall study population (3.1 log10 copies/mL) through week 48. There was no significant difference in the CD4 cell count at baseline and the increase in the CD4 cell count, through week 48, among the HIV-HBVcoinfected patients who received tenofovir DF (151 cells/mm3 and 99 cells/mm3, respectively) and stavudine (249 cells/mm3 and 142 cells/mm3, respectively). There also was no significant difference in the increase in the CD4 cell count, through week 48, among the HIV-HBVcoinfected patients with (147 cells/mm3) or without (81 cells/mm3) grade 3 elevations of ALT.
DISCUSSION
Among individuals coinfected with HIV and HBV in 2 randomized controlled trials, therapy with tenofovir DF has clearly demonstrated anti-HBV virologic efficacy. During 48 weeks of therapy with tenofovir DF, a mean reduction of 4 5 log10 copies/mL in the HBV DNA level was seen in antiretroviral therapyexperienced HIV-HBVcoinfected individuals with or without resistance to lamivudine. During the 48 weeks of the study, a similar reduction in the HBV DNA level was seen in antiretroviral therapynaive HIV-HBVcoinfected individuals who received combination therapy with lamivudine and tenofovir DF as a component of their initial 3-drug HAART regimen. A trend toward greater suppression of HBV DNA as well as reduced YMDD resistance in HIV-HBVcoinfected individuals who were receiving lamivudine and tenofovir DF, compared with lamivudine alone, were additional findings. One interpretation of the results for antiretroviral therapynaive individuals is that dual anti-HBV active antiretroviral therapy may provide improved outcomes for HIV-HBVcoinfected populations. However, it is notable that the mean reduction in HBV DNA levels was similar in treatment-experienced individuals with lamivudine resistance who were receiving tenofovir DF and in treatment-naive individuals who were receiving both lamivudine and tenofovir DF.
Some limitations of our study methodology should be considered in the interpretation of the findings. Both substudies were protocol defined among study participants enrolled in large randomized controlled trials for the assessment of the safety and efficacy of the use of tenofovir DF for HIV infection. The randomized nature of these studies is an advantage; however, the relatively small sample size of both substudies limited the power to demonstrate significant outcomes. Specific information on HBV-related disease, such as liver biopsy findings, was not obtained. However, a recent review has shown a high correlation between suppression of HBV DNA and histologic improvement in patients receiving anti-HBV treatment. The exclusion, from both study 903 and study 907, of patients with high serum levels of transaminases means that our findings are unable to be generalized to individuals with HIV-HBV coinfection and high levels of hepatic inflammation. However, previous studies of HBV therapy have shown a correlation between higher ALT levels and improved therapeutic outcomes. HBeAg seroconversion has been reported in HIV-HBVcoinfected patients after commencement of non-HBV active antiretroviral therapy, with immune restoration considered to be the explanation for the seroconversion. In both substudies, there was no significant difference in the increase in the CD4 cell count in the tenofovir DF arm versus the comparison arm. Thus, immune restoration would appear to be unlikely as an alternative explanation for the improved efficacy of tenofovir DF.
The virologic efficacy of tenofovir DF for antiretroviral therapyexperienced HIV-HBVcoinfected individuals is consistent with that noted in previous open-label studies. The largest of these previously published studies followed, for 52 weeks, 20 HIV-HBVcoinfected individuals who were receiving tenofovir DF. The reduction in the HBV DNA level (4 log10 copies/mL) and the equivalent efficacy of tenofovir DF against wt HBV and lamivudine-resistant HBV in that study were similar to our findings. The equivalent efficacy of tenofovir DF against wt HBV strains and lamivudine-resistant HBV strains is consistent with the findings of previous in vitro studies. The vast majority of individuals in both of our substudies were HBeAg positive. However, a recent report has also demonstrated rapid reductions in HBV DNA after commencement of tenofovir DF therapy for 5 HIV-HBVcoinfected HBeAg-negative individuals with lamivudine resistance.
The efficacy of tenofovir DF therapy for individuals with lamivudine-resistant HBV provides a means to prevent or control breakthrough and withdrawal hepatic flares seen in HIV-HBVcoinfected individuals with YMDD mutant strains. Recent evidence suggests that YMDD mutations may also provide a means for escape of HBV vaccine. Thus, tenofovir DF could potentially reduce transmission of resistant HBV to both vaccinated and unvaccinated contacts.
Similar suppression of HBV DNA in antiretroviral therapyexperienced HIV-HBVcoinfected individuals has been demonstrated with the use of adefovir dipivoxil. Although adefovir dipivoxil has demonstrated antiHIV-1 activity at doses of 60120 mg/day, nephrotoxicity has prevented it from further development as an antiretroviral therapy agent. The 10-mg dose of adefovir dipivoxil that has demonstrated efficacy in HBV-monoinfected and HIV-HBVcoinfected populations has no significant anti-HIV activity. In contrast, the dual HBV and HIV-1 activity of tenofovir makes it an ideal agent for the treatment of HIV-HBVcoinfected individuals.
Compared with monotherapy, combination antiviral therapy has clearly demonstrated improved efficacy for the management of HIV infection and chronic hepatitis C virus infection. However, there has been relatively limited research regarding the use of combination antiviral therapy for chronic HBV infection. Regimens that have been examined include lamivudine and interferon- as well as lamivudine and famciclovir, with limited or no improved efficacy, compared with monotherapy. In HBV-monoinfected populations, other studies of combination therapy, including lamivudine and -Lthymidine, as well as lamivudine and adefovir dipivoxil, are under way, and a study of HBV viral kinetics suggests improved efficacy with combination antiviral therapy. The study 903 substudy is the first comparison of lamivudine with lamivudine plus tenofovir DF in antiretroviral therapynaive HIV-HBVcoinfected individuals. Trends toward greater suppression of HBV DNA and reduced YMDD mutations in the dual-therapy arm of the study provide preliminary evidence that this particular combination may be more effective than lamivudine monotherapy.
However, there are several questions that need to be answered in larger prospective randomized clinical trials. These include the relative potency of combination lamivudine and tenofovir DF, compared with both lamivudine monotherapy and tenofovir DF monotherapy, and maintenance of HBV DNA suppression and longer-term resistance in those receiving combination lamivudine and tenofovir DF therapy. The effect of combination therapy on HBeAg seroconversion and progression of liver disease also needs to be examined in larger studies with more prolonged follow-up.
|
|
| |
| |
|
|
|