 |
|
|
| |
Antiretrovirals and other stuff!
|
| |
| |
Dublin 10th EACS 2005
Report for www.NATAP.org
Reported by Mike Youle, MD, Royal Free HIV Clinic, London, UK
Contribution/Editing by Jules Levin
Cold fair Dublin in the far West of Europe had its best face on for this 10th European AIDS Clinical Society Conference, the sun shone and the conference attracted a large number of delegates from across the entire sub-continent. The quality of the presentations was high and the meeting continued to be a cross between education and the showing of new data for both treatment and care.
So here is my usual round up of new agents presented at the meeting:
This was a stormy month for the CCR5 inhibitors with both vicriviroc from Schering-Plough and aplaviroc from GlaxoSmithKline (GSK) running into trouble. The last of the three agents in clinical trials with a clean sheet is maraviroc developed by Pfizer, although a single case of severe liver toxicity has been reported in a patient, it appears that this is much more likely to be related to concomitant medication with known liver-toxicity. Of the two presentations on the drug at the EACS meeting the first was of in vitro interaction data between maraviroc (formerly UK427,857) and 17 other approved antiretrovirals, including enfurvitide [PE7.1/2]. Additive interactions were seen with most drugs (14/17) with synergy noted for atazanavir, indinavir and enfurvitide but on further testing on an additive effect was seen there also; importantly no antagonism was found. The second study confirmed that although some postural hypotension (drop in blood pressure on standing) was reported in the early studies this agent does not have a significant effect on cardiovascular function, [LB 4.1/10].
Non-nucleoside reverse transcriptase inhibitor (NNRTI) data at the conference included pharmacokinetic information for TMC278 (dosed at 150mg qd) and lopinavir/ritonavir in healthy volunteers [PE4.3/1]. When combined the TMC278 area under the curve (AUC) was 152% and the Cmax and Cmin were 129% and 174% respectively whereas the levels of lopinavir/ritonavir were unaltered suggesting that CYP3A4 inhibition had boosted the levels of TMC278. There is a phase IIB study currently underway which will decide the recommended dose.
The other Tibotec NNRTI, TMC125, is further along in development and Jeff Nadler and Julio Montaner presented back-to-back studies in the late-breaker sessions describing the findings in two medium sized trials in treatment experienced individuals [LBPS3/7A, LBPS3/7B]. The first, TMC125-C223, was a randomised dose finding study, run in the USA in 199 subjects with documented resistance to NNRTI’s and >3 PI primary mutations. Subjects either received TMC125 (400mg or 800mg) with an optimised background, or optimized regimen alone. Median VL at baseline was 4.7log and T4 around 100 cells. Baseline resistance patterns are shown below as well as the viral load outcomes:
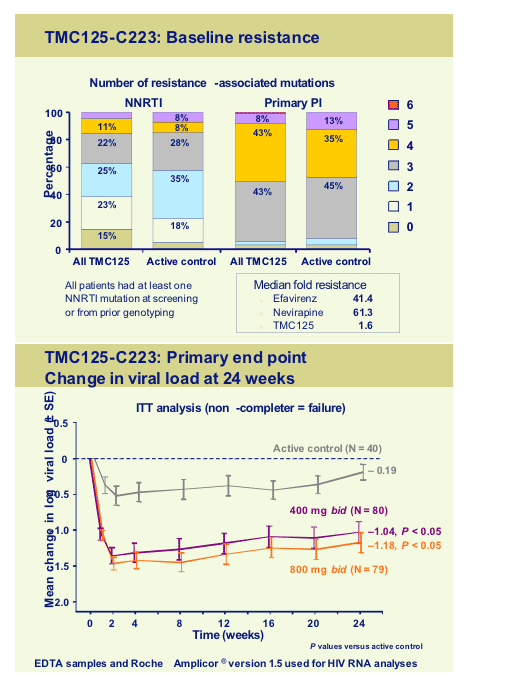
In terms of adverse events there seemed to be little additional toxicity in the two TMC125 arms although reported rash of any aetiology was reported in 20% of TMC patients but only in 8% of controls, however only 2% was thought to be related to drug. This seems markedly better safety profile than with the current available NNRTI’s.
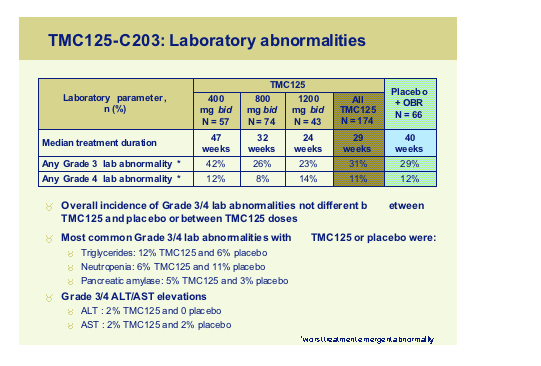
The second study TMC125-C203 is a 48 week randomised, blinded, placebo controlled dose-escalation trial in 3 class experienced patients:
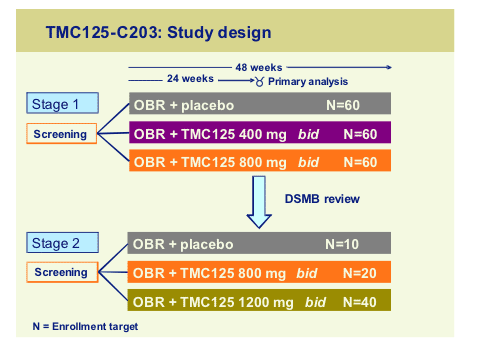
The study was planned to assess the possibility to increase the dose of TMC125 up to 1200mg bid. All subjects received an optimized background regimen with at least two active drugs and this was reflected in the viral load outcomes in Stage 1 where all three arms achieved similar viral load declines.
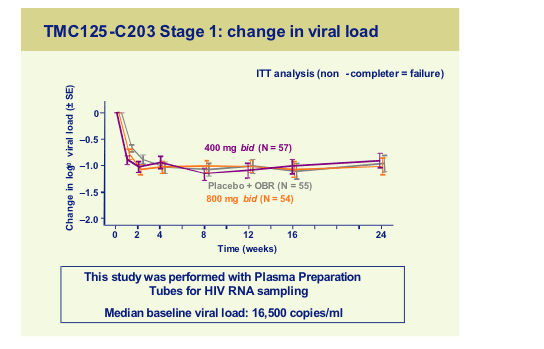
When it came to toxicity there appeared to be no consistent differences between the TMC125 arms at any dose and the control arm with regard to rash, liver function abnormalities, pancreatitis and CNS toxicity once again confirming that this agent probably has fewer of the disadvantages inherent with the current NNRTI’s.
Probably the most exciting presentation of the whole meeting was that given by Javier Morales-Ramirez on the integrase inhibitor from Merck; MK0518 [LBPS1/6]. MK-0518 is an agent that prevents HIV from incorporating its genetic information into that of the host cell and thereby prevents established infection of T cells. It has shown good efficacy in vitro and is synergistic with many available HIV drugs and since it has a novel mode of action, within the nucleus of the cell, there should be no cross resistance and thus represents a new option for those with multi-drug resistance. The good news continued with no safety issues from early studies and good tolerability and no requirement to dose with food.
In this randomized, double-blind, placebo-controlled study twenty eight treatment-naïve patients received 10 days of monotherapy with MK-0518 (at doses of 100mg, 200mg, 400mg or 600mg) and 7 study participants received placebo. HIV viral load reductions were a potent -1.7 to 2.2 log for the various doses of the integrase inhibitor administered. 50-57% of patients receiving MK-0518 achieved <400 copies/ml and 13-29% achieved <50 copies/ml viral load. The drug appeared safe and well tolerated with no serious adverse events and no discontinuations due to adverse events. .
Mean HIV RNA across the groups was 4.53 to 4.97log and mean CD4 count: 256-569. No drug related adverse events were seen of any consistency. At day 10 viral load reductions ranged from -1.7 to 2.2log for individuals receiving MK-0518.

Overall these are very encouraging results and a Phase 11 dose-ranging study is underway against efavirenz. Watch this space!
GW640385 is a novel aspartyl protease inhibitor (PI) being developed by GSK and about to enter Phase 2 trials. An in vitro study against a panel of multiple resistant isolates (containing mutations at positions 32, 33, 46, 47, 50, 54, 82, 84 and 90) was presented by Yates and showed that compared to the current PI’s, TMC114 and tipranavir were not included, this agent showed good effect when cultured with these isolates [PE3.3/3]. Only two virus showed >5 fold resistance to this drug and 11 with >2.5 fold changes. GW640385 was consistently the most potent of the PI’s tested but no signature mutation or pattern emerged in this study and it will be to the clinical studies that we look for more information of how this drug will perform.
A number of new presentations on tipranavir (TPV) were shown consisting mainly of follow-up of the large RESIST studies.
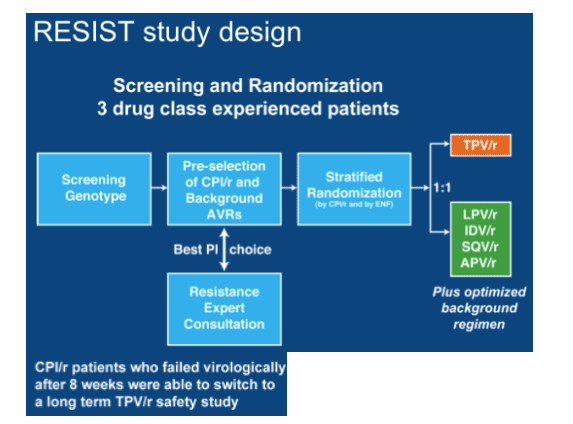
That of Cassetti from Buenos Aires focused on those who in the two large studies had reached the treatment response of > 1log reduction in viral load at week 24 (240/582 [41.2%] and mapped what happened to them [PE2.7/1]. Of these 79.6% reached <400copies/ml and 55.8% (134/240) achieved <50copies/ml and the median viral load drop was 2.6log, with 75% getting at least 1.8log reduction. Median T4 cell rise was 88 cells in responders compared to 11 in non-responders. Moreno showed further data on the use of genotypic sensitivity score (GSS) in the RESISTS studies using the Virtual Phenotype or Trugene assays [PE3.3/4] which showed that the percentage responding to tipranavir increased as the GSS score became greater. One of the problems with this analysis is that the exact link between particular combinations of mutations and resistance to tipranavir remains somewhat uncertain so that this is at best a guide to the likelihood of success with the drug.
One way of improving the use of resistance tests was proposed within a poster from Brendan Larder who suggested that neural networks are more accurate predictors of viral response than rules-based genotypic interpretation systems [PE3.4/13]. He used artificial neural networks (ANN) trained to predict change in viral load from 2,983 treatment change episodes using input variables of 55 resistance mutations, drugs in the new regimen, baseline viral load and time to follow-up. Using this system compared to the ANRS, Stanford and Rega rules based systems ANN were more accurate at predicting outcome explaining 75% of the variation compared to no more than 29% for the other systems and suggesting that this may be a way forward as the resistance patterns become ever more complex.
Another cut of the RESIST data surfaced in a poster by Charles Farthing which stratified outcome by viral load and T4 response [PE7.3/22] suggesting that neither of these parameters predicted a poor outcome and that all subgroups did better with tipranavir as a new PI.
--TPV/r provided superior efficacy to CPI/r regardless of baseline VL and CD4+ cell count
--TPV/r improved virologic response even in patients with high baseline viral
loads (>100,000 copies/mL)
--Patients in the TPV/r arm with low baseline CD4+ cell counts (<50 cells/mm3) had an improved treatment response compared with those in the CPI/r arm
--In treatment-experienced patients, TPV/r provides a good treatment
response in patients with both high and low viral loads
For patients with <10,000 c/ml at baseline 56% had treatment response with TPV/r vs 31% for CPI/r; 41.7% of patients with viral load of 10,000 to 100,000 c/ml who took TPV/r achievd a treatment response compared to 19% taking CPI/r; and for patients with >100,000 c/ml at baseline 35% taking TPV/r vs 13.7% taking CPI/r achieved a "treatment response". As you can see in table response to TPV/r was the same regardless of whether baseline CD4 count was >350, 200-350, or 50-200. When CD4 count at baseline was <50 the treatment response was less for patients taking TPV/r, but it was still superior to patients who received CPI/r (26% vs 9%).
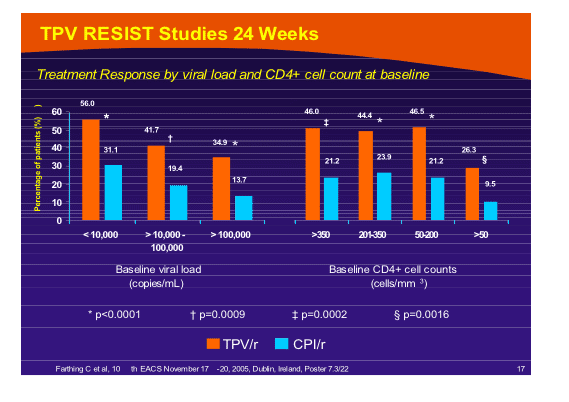
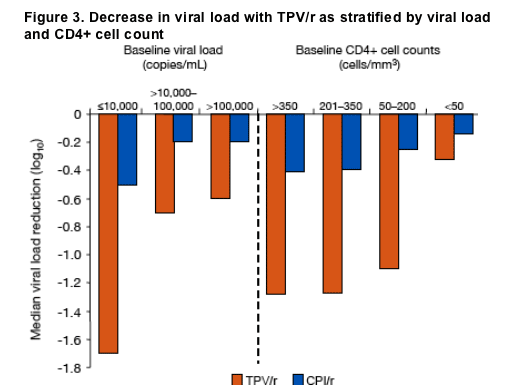
Figure 3. Decrease in viral load with TPV/r as stratified by viral load
and CD4+ cell count
Yet more slicing produced the outcome reported by Jurgen Rockstroh that when stratified by previous PI use the 24 week outcomes no diminution of “treatment response” was seen when tipranavir was introduced [PE7.9/11]. When evaluating <400 copies/ml response anf by reduction in viral load, previous PI did appear to matter.
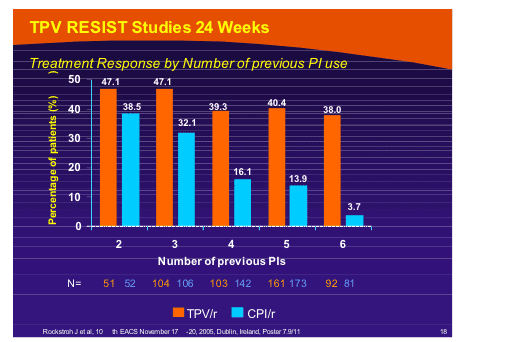
--The overall treatment response (≥1 log10 reduction in 2 consecutive viral load measurements) for patients in the TPV/r arm was 41.2% (240/582) vs 21.3% (109/577) in the CPI/r arm
--Patients in the TPV/r arm had a consistently improved treatment response compared to the CPI/r arm in all PI strata
--Almost 50% of TPV/r patients with 3 or fewer previous PIs achieved a treatment response
--The difference in treatment response became more dramatic as the PI experience of patients increased
--In those with 6 previous PIs, TPV/r had a 10 times greater treatment response than in CPI/r patients (38.0% [35/92] for TPV/r vs 3.7% [3/81] for CPI/r)
--Approximately 40% of TPV/r patients presenting with 4 to 6 previous PIs at baseline achieved a treatment response
Virologic response (<400 copies/mL and <50 copies/mL)
at 24 weeks stratified by previous PI use
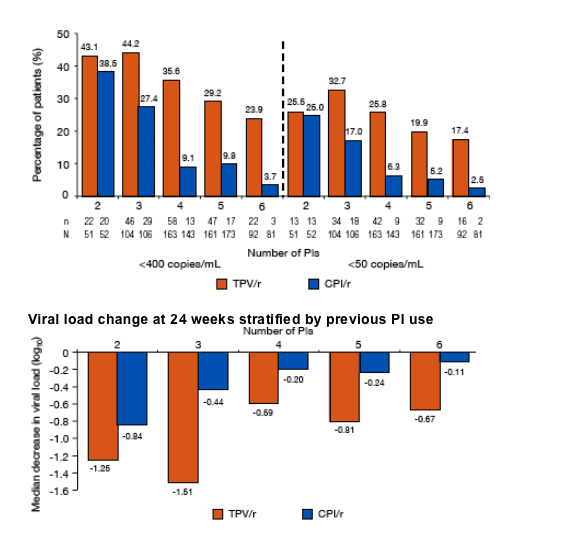
Viral load change at 24 weeks stratified by previous PI use
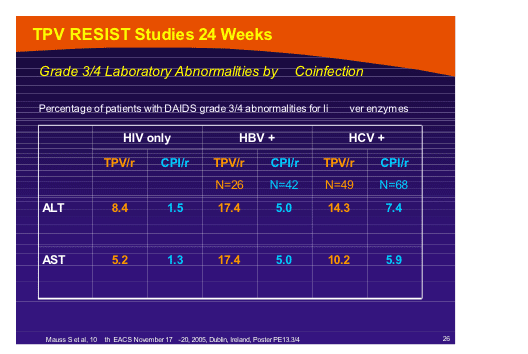
In addition data that showed some increase in liver-related laboratory abnormalities in patients who were co-infected with HBV (3.5% of the study population) or HCV (6.6%) was shown by Stefan Mauss [PE13.3/4]. How this will translate into clinical practice and to what degree it is due to the higher doses of ritonavir used compared to other PI’s will require information from the ongoing studies of tipranavir/ritonavir (500/100mg bid).
After all these posters the meatiest data came in an oral presentation by Pedro Cahn who showed the 48 week meta-analysis of the RESIST 1 and 2 studies [LBPS3/8]. At last some data separated out the effect of enfurvitide (T20) from tipranavir within the study although Boehringer-Ingelheim seem still coy about showing the data of tipranavir versus tipranavir plus T20 for fear of isolating their agent to the salvage situation, a view seemingly held by marketers rather than scientists. The subgroup who received both drugs as new agents did much better and since tipranavir is limited in which antiretrovirals it can be dosed with (although data now exists showing no significant pharmacokinetic interaction with maraviroc [LBPE4.3/15]) it would seem a wise course of action to use as many active drugs together as possible rather than burning through each agent as mono- or sub-optimal therapy.
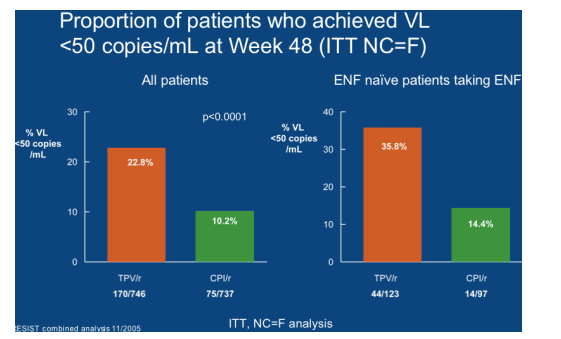
Although reasonably well tolerated there were greater AST/ALT and triglyceride changes in the tipranavir arms and it remains to be seen if the reduction of ritonavir to 100mg bid in the naïve studies will translate into fewer abnormal values.
More data was shown on TMC114 including a pharmacokinetic study to assess the effect of food on the levels achieved of the 400mg tablet formulation of TMC114 in 24 healthy volunteers [PE4.1/1]. In the presence of low-dose ritonavir systemic exposure of TMC114 was increased by around 30% after a meal compared to on an empty stomach suggesting that the type of meal is relatively unimportant but that the drug should be given with food.
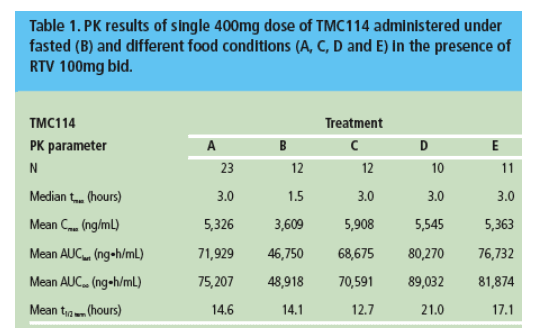
Further pharmacokinetic data in healthy volunteers mapped the effect of combining TMC114 with atazanavir both in combination with low-dose ritonavir [PE4.3/4]. Twenty three subjects with the single agents boosted or a combination of TMC114/ritonavir (4400mg/100mg bid) plus atazanavir (300mg qd). Whilst TMC114 levels were unaffected by the addition of the second PI, the atazanavir pre-dose levels rose by 87% and the exposure to ritonavir was increased by 50%. The drugs can be combined but clinical studies will be required to see if any adverse events result from the higher drug levels.
Clinical data on this new PI was provided by Montaner and co-workers who reanalysed the POWER studies to evaluate the potential clinical and mortality endpoints using the week 24 CD4 and viral load data via two methods of either regression using a meta-analysis of randomised clinical endpoint studies conducted before the introduction of HAART or categorisation using data sets derived from cohort studies of CD4 and clinical progression rates in HAART treated patients [PE18.4/4]. The additional effect of TMC114/r over the comparator PI/r in raising CD4 levels and suppressing VL were predicted to lower progression rates to AIDS or death by 48% using the categorisation method and 55% with the alternative regression analysis. The use of these analyses has become ever more helpful since it is no longer ethical or practical to conduct large scale clinical endpoint studies in late stage HIV disease.
Jose Arribas showed an updated set of information on the OK study in which he evaluated lopinavir/ritonavir (Kaletra) as monotherapy in patients who had never failed a PI and were suppressed to <50 copies for more than six months [PE7.5/5]. After 72 weeks the percentage of individuals at VL <50 (ITT-M=F) was 81% in the monotherapy group and 90.5% in the continue HAART group (p=0.38). There were 3 transient blips in the monotherapy arm and two in the triple therapy arm whilst 4 subjects had viral failures whilst on Kaletra which was re-suppressed by the re-addition of the nucleosides. A much larger study is ongoing in Spain which will more clearly define the risks and benefits to this approach. Finally a cost-effectiveness study has been performed within the OK study [PE19.5/2] which showed a cost-effectiveness ratio for Kaletra monotherapy of 5,186 Euros per unit of effect achieved (VL<50 at week 48) compared to 8,688 Euros for the standard triple therapy arm, suggesting that monotherapy could be more cost-effective as an intervention.
Tabatha Mahungu presented data on all HIV-infected patients attending the Royal Free Centre for HIV Medicine who had received SQV-500 in a retrospective cohort study from September 2004 until October 2005 [PE1.1/5]. 137 adults were enrolled taking SQV-500-containing regimens that included SQV/r 1000/100 mg bid (n = 116) or 2000/100 mg qd (n = 19) and followed for a median of 275 (range 136–513) days.
Kaplan-Meier estimates showed that 95.6% (95% CI: 92.1, 99.0) of study subjects remained on SQV-500 therapy at 24 weeks and no new AIDS diagnoses or deaths were observed. Eighty four percent of the 92 subjects with <50copies/mL at baseline remained undetectable throughout whilst 15 subjects had one blip above this level during follow-up; no subject had sustained viraemia whilst taking therapy.
To assess the impact of SQV-500 therapy on LFT and lipid markers, measurements before and after initiation of SQV-500 therapy were and no adverse changes in these parameters were noted.
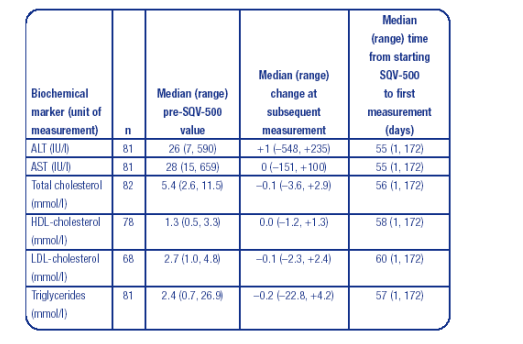
The results of this 24-week retrospective cohort analysis provide an encouraging first insight into how SQV-500 performs in clinical practice, with the majority of patients tolerating the formulation well and maintaining favourable virological and immunological responses.
After all the discussion about the role of the interaction between PI’s and agents that reduce stomach acidity a study from Alan Winston at least showed that saquinavir has a favourable profile with omeprazole in subjects dosed with the 500mg formulation of saquinavir [LB4.3/16]. Eighteen healthy volunteers were dosed with saquinavir 1000mg/ ritonavir 100mg twice daily for 15 days and from day 11 to15 were co-administered omeprazole 40mg qd.
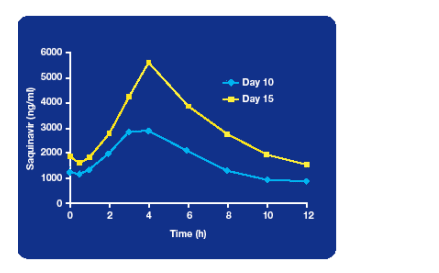
In the presence of omeprazole total saquinavir AUC was increased by 82% but no short-term toxicity was seen and the levels reached were similar to those seen in other studies with standard doses of saquinavir suggesting that this change is not likely to lead to an increase in side effects.
There was some extra information on Interleukin 2 provided from the GESIDA study from Spain in which they assessed 162 patients who had at least 1 year on HAART and had a low level (<5,000 copies/mL) of viraemia or were undetectable (87%), [PS3/1]. Subjects had not had a CD4 rise >200 cells/_l and median CD4 at inclusion was 137. Median CD4 after a third cycle rose to 211 (IQR123-289) (p<0.0001) and 88.4% were undetectable, whilst 44% had a CD4 >200, allowing discontinuation of opportunistic infection prophylaxis. This study strengthens the data originally presented in the French ILSTIM study several years ago and suggests that this is a sensible and viable intervention for those with poor immune reconstitution. We still await the findings of the multinational SILCAAT study.
An interesting approach to the assessment of adherence was taken by Giola and co-workers in Varese, Italy where they evaluated 60 consecutive out-patients to evaluate in a blinded fashion if there was link between sub-optimal adherence and personality [PS3/2]. Previous work has suggested that health beliefs and interaction with health care providers is affected by personality but this is the first work in this area on HIV patients, as far as I am aware. Subjects filled in a validated personality test (Cattel’s 16PF) as well as a standardised questionnaire regarding their attitudes to HAART and the psychologist assessing the test was masked as to the therapy of the individuals. Twenty eight were on an MMRTI-based regimen and 32 on PI-based therapy and 11 (34%) of the former group and 6 (21%) of the latter group were judged to be poorly adherent by the clinicians involved in their care. Underlying personality disorders such as low ego strength and poor compulsion control as well as depression and anxiety were strongly associated with poor adherence, with all 17 of the group who had been designated as poor adherers having one of more personality disorders whilst the good adherers showed none of these traits (p<0.001). This study is important since whilst it is not possible to alter your underlying personality it is possible to modify the way people act within this framework.
Thank you for those investigators who shared slides from their presentations.
Mike Youle
|
|
| |
| |
|
|
|
|
|