 |
|
|
| |
The Pharmacokinetic Interaction Between
Fosamprenavir/Ritonavir and Atazanavir
in Healthy Adult Subjects (APV10018)
|
| |
| |
The 10th European AIDS (EACS) Conference
17-20 November 2005, Dublin, Ireland
Poster Number PE4.3/9
M.B. Wire1, M.J. Shelton1, Y Lou1, S. Agarwala2, M. Child2, S.S Min1
1GlaxoSmithKline, Research Triangle Park, NC, USA; 2Bristol-Myers Squibb, Princeton, NJ, USA
Fosamprenavir (FPV, Lexiva, Telzir), the phosphate ester pro-drug of amprenavir (APV), and atazanavir (ATV, Reyataz) are both HIV-1 protease inhibitors (PIs) approved for the treatment of HIV infection in adults. Both FPV and ATV may be administered with ritonavir (RTV), which boosts plasma APV and ATV concentrations primarily via inhibition of CYP3A4.
Combination dual RTV-boosted HIV-1 PI therapy is used in the clinical management of patients, especially in treatment-experienced patients who have few remaining treatment options. Because APV, ATV, and RTV are all CYP3A4 substrates and inhibitors and because APV and RTV also have CYP3A4 induction properties, the impact of combining all three HIV-1 PIs on plasma APV and ATV PK was unknown. Therefore, this study evaluated the pharmacokinetic (PK) interaction between FPV/RTV BID and ATV QD.
Study Design
This was a randomized, open-label, two-period, two 2x2 cross-over study. Healthy adult subjects underwent screening assessments within 30 days of dosing to determine their eligibility for enrollment into the study. Enrolled subjects remained at the study center throughout each period, where they received study drugs as described in Table 1.

Key Entry Criteria
Healthy adult (18-55 years) male and female subjects with BMIs between 18.5 and 29.9kg/m2 who were not taking concurrent medications were eligible for the study. Subjects were excluded if they had ALT, AST, or bilirubin levels above the upper limit of the reference range or a history of first degree or higher atrioventricular (AV) block or if cardiac abnormalities were identified on the screening ECG.
Dosage & Administration
The study drugs and dosages administered to subjects are described in Table 1. All morning doses (which included all ATV doses) were administered within 15 minutes after completion of a light breakfast (approximately 400kcal, 10g fat,
and 10g protein). All evening doses were administered within 15 minutes after completion of supper.
Safety Assessments
Adverse event and concurrent medications were collected throughout the study. Full clinical laboratory assessments were done at Screening, Days 1, 4, and 10 and at follow-up; additionally, limited clinical chemistry testing, including
AST, ALT, alkaline phosphatase, GGT, total bilirubin, and fractionated bilirubin, was done on Days 6 and 8. Vital signs were done at Screening, Days 1, 4, and 10 and at follow-up; ECGs were also done at Screening (all treatments) and on
Days 1, 4, and 10 for the ATV/RTV and FPV/RTV + ATV treatments.
Pharmacokinetic Assessments
Pre-dose samples were collected on Days 4, 6, 8, 9 and 10 of each period. Serial PK samples were collected on Day 10 of each period at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, and 12 hours after dosing; additional samples were
collected at 16 and 24 hours after dosing for the ATV-containing treatments.
Plasma APV and RTV concentrations were measured by GlaxoSmithKline and ATV concentrations were measured by Bristol-Myers Squibb. All analyte concentrations were determined by high performance liquid chromatography with
tandem mass spectrometry (HPLC/MS/MS).
Pharmacokinetic & Statistical Analysis
Plasma PK parameters were derived by noncompartmental methods. Analysis of variance (ANOVA), considering arm, period, and treatment as fixed effects and subject within arm as a random effect, was performed using SAS Mixed
Linear Models procedure to compare plasma APV, RTV and ATV PK between the treatments in two separate models.
The model for plasma APV and RTV PK comparisons included Arms 1 and 2 and the model for plasma ATV and RTV PK comparisons included Arms 3 and 4. The ratio of geometric least squares (GLS) means and associated 90% CIs were estimated for plasma APV and RTV PK treatment comparison FPV/RTV + ATV vs FPV/RTV and plasma ATV and RTV treatment comparisons FPV/RTV + ATV vs ATV/RTV.
Subject Accountability
Eighty-six subjects were enrolled and included in the safety analyses. The safety population was 81% male and 19% female, 71% White, 13% Black, 5% Asian, 3% Hispanic, and 8% of other race. The median (range) age was 26 years
(18-55 years), height was 173cm (151-192cm), weight 74.6kg (53.2-96.3kg), and body mass index 24.8kg/m2 (18.7- 30.7kg/m2).
Forty-three subjects completed the study and were included in the statistical analysis of the PK data; 22 subjects were included in the APV treatment comparisons and 21 subjects were included in the ATV treatment comparisons.
Forty-five subjects prematurely discontinued from the study: 22 subjects (26%) withdrew due to adverse events; 11 subjects withdrew due to “other” reasons; 10 subjects withdrew consent to participate in the study; and 2 subjects were
lost to follow-up. “Other” reasons for premature discontinuation included: employment, concern for side effects of study medication, family emergency, positive urine drug screen, inability to swallow RTV, child care issues, no show for Period 2, and inability to get time off of work.
RESULTS
The PK results of this study showed that co-administration of FPV 700mg BID + RTV 100mg BID with ATV 300mg QD had no effect on steady state plasma APV PK. These results are in agreement with previous observations that the effect of RTV on APV exposure was larger than the effect of ATV on APV exposure.1
Co-administration of FPV 700mg BID + RTV 100mg BID with ATV 300mg QD decreased plasma ATV AUC(0-_) by 22% and Cmax by 24%, but C_ remained unchanged, compared to co-administration of ATV 300mg QD with RTV 100mg QD. These results are consistent with the results of a study where co administration of ATV 400mg QD with APV (delivered as Agenerase) 1200mg QD decreased plasma ATV AUC(0-_) by 31% and Cmax by 41%.2
When ATV 300mg QD was added to FPV 700mg BID + RTV 100mg BID plasma RTV AUC(0-_) was increased by 93%, Cmax by 96%, and C_ by 37%. In another study, co-administration of ATV 300mg QD with saquinavir 1600mg QD + RTV 100mg QD also increased plasma RTV exposures, but to a lesser extent; plasma RTV AUC(0-_) increased by 40% and Cmax by 58%, whereas C_ decreased by 27%.3
There was a high withdrawal rate (53%) from this study; however, this did not appear to impact the PK results because--
1) pre-dose plasma APV and ATV concentrations collected prior to premature withdrawal appeared similar to pre-dose concentrations collected from subjects who completed the study (data not shown), and 2) the study was conducted in a
cross-over fashion such that subjects included in the statistical analysis contributed data for both test and reference treatments.
The types of AEs and the AE rates were similar to those reported for FPV/RTV and ATV/RTV in previous studies and the discontinuation for AE rates were similar to those reported for FPV/RTV in previous studies. No significant changes
in cardiac conduction were noted with the combination of FPV/RTV and ATV.
AUTHOR CONCLUSION
Given no change in plasma APV exposure, a modest decrease in plasma ATV exposure, and no new safety issues for the combination, the dual RTV-boosted PI regimen of FPV 700mg BID + RTV 100mg BID + ATV 300mg QD could be further evaluated in clinical trials in HIV-infected patients.
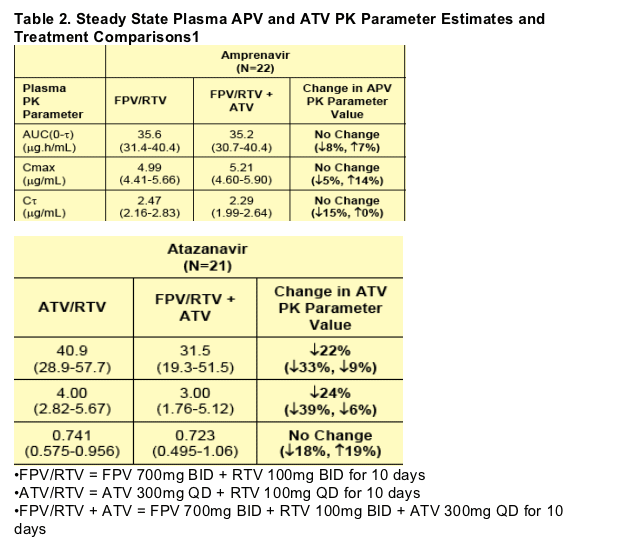
1.Geometric Mean (95% CI) presented for each PK parameter; Percent Change (90% CI) presented for each treatment comparison.
When ATV 300mg QD was added to FPV 700mg BID + RTV 100mg BID, plasma RTV AUC(0-_) was increased by 93%, Cmax by 96%, and C_ by 37%.
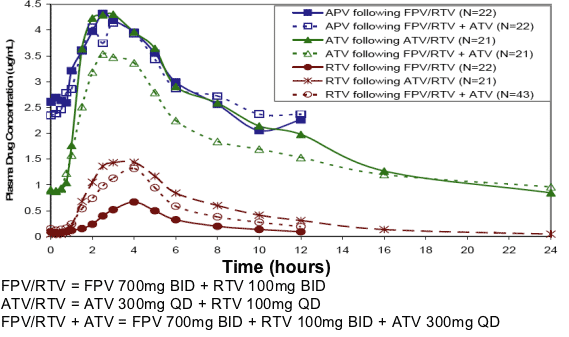
Figure 1. Median Plasma APV, ATV, and RTV Concentration-Time Profiles by Treatment
Safety
Adverse Events
Twenty-two of 86 subjects (26%) were withdrawn from the study due to AEs. Rates of subject withdrawal due to AEs by treatment were FPV/RTV: 7/31 (23%), ATV/RTV: 2/33 (6%), and FPV/RTV + ATV: 13/72 (18%). The most common
AEs leading to withdrawal were rash: 14/22 (64%) and vomiting: 2/22 (9%). All other AEs leading to withdrawal, including nausea, toothache, fatigue, non-cardiac chest pain, chest wall pain, headache, insomnia, and dyspnea, occurred in one subject each. Premature subject withdrawal due to rash occurred in the FPV-containing treatments only.
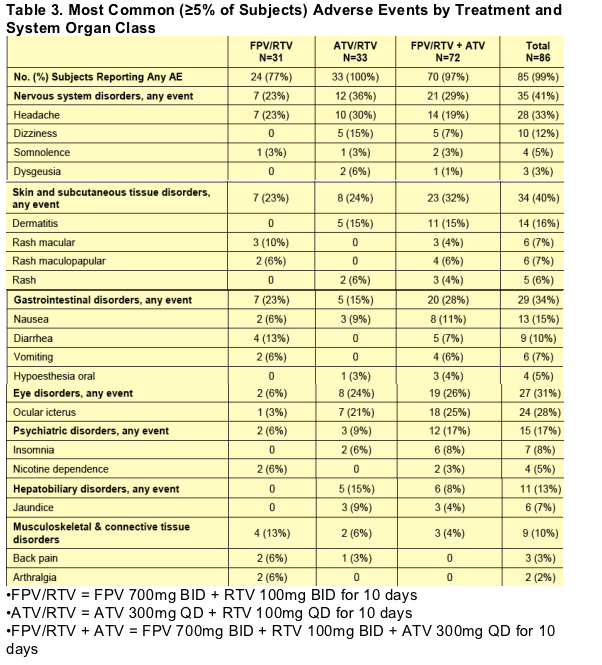
For AEs reported by at least 5% of the subjects, frequencies are summarized by treatment in Table 3. The most frequently reported AEs were headache (33%), ocular icterus (28%), dermatitis (16%), and nausea (15%). A higher percentage of subjects in the ATV-containing treatments [ATV/RTV (100%) and FPV/RTV + ATV (97%)] reported overall AEs compared to those receiving FPV/RTV (77%).
Drug-related skin and subcutaneous tissue disorders were reported in 28% of subjects overall, 19% of subjects receiving FPV/RTV, 6% of subjects receiving ATV/RTV, and 22% of subjects receiving FPV/RTV + ATV. The majority
of drug-related skin and subcutaneous tissue disorders were rash; most reports of dermatitis were related to contact with the ECG electrodes and were not considered drug-related.
Table 3. Most Common (≥5% of Subjects) Adverse Events by Treatment and System Organ Class
Four subjects experienced severe adverse events, including severe gas pain (1 subject) and severe headache (3 subjects). One subject experienced a non-fatal serious adverse event of abnormal computed tomography (CT) scan
of the head; the investigator considered that there was no reasonable possibility that the event was related to investigational products.
Clinical Laboratory Tests
The most commonly reported clinically significant drug-related laboratory abnormality, as assessed by the study investigator, was increased bilirubin (unconjugated) in 73/86 subjects (85%) overall, including 1/31 subject (3%) receiving FPV/RTV, 31/33 subjects (94%) receiving ATV/RTV, and 68/72 subjects (94%) of subjects receiving FPV/RTV + ATV. At any given time point during ATV/RTV dosing, up to 46% of subjects had Grade 3 and up to 11% had Grade 4 total bilirubin values. At any given time point during FPV/RTV + ATV dosing, up to 50% of subjects had Grade 3 and up to 8% had Grade 4 total bilirubin values.
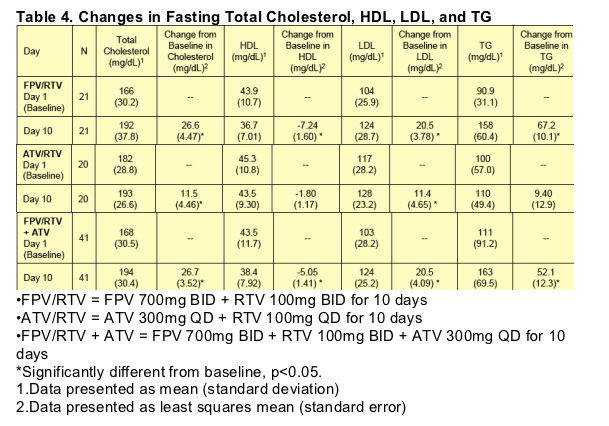
No Grade 3 or 4 ALT or AST values were reported during the study. Grade 1 and 2 ALT and AST values were observed on the combination FPV/RTV + ATV treatment only; four subjects had Grade 1 ALT, one subject had Grade 2 ALT, and four subjects had Grade 1 AST on-treatment values. One subject had a Grade 1 ALT value at follow-up (without ALT abnormalities on treatment), and one subject had Grade 1 ALT, Grade 2 AST, and Grade 4 CPK values observed at follow-up that were considered due to strenuous exercise. Two subjects had Grade 3 neutropenia on the last day of FPV/RTV + ATV dosing. Changes in fasting lipids are presented by treatment in Table 4.
ECG
No clinically significant changes in vital signs, QTcF, or PR interval were observed during this study. No subject had a change from baseline in QTcF of greater than 60msec or a QTcF value greater than 500msec while receiving FPV/RTV+ ATV. Four subjects had ECG or telemetry abnormalities that were reported as AEs. Two subjects receiving ATV/RTV were reported to have T wave inversion and ventricular extrasystoles, and two subjects receiving FPV/RTV +
ATV were reported to have ventricular and supraventricular extrasystoles. These ECG changes were not considered clinically significant.
|
|
| |
| |
|
|
|
|
|