| |
FDA Antiviral Drug Advisory Committee Recommends Unanymously to Approve Entecavir for Hepatitis B
|
| |
| |
Today March 11, the FDA Antiviral Drug Advisory Committee met in Gaithersburg, Maryland to hold a public hearing to consider the approval of entecavir for treatment for hepatitis B chronic infection. The brand name for entecavir is Baraclude.
Regarding HIV, BMS reported that entecavir does not have anti-HIV activity. There does not appear to be risk for drug-drug interactions with antiretrovirals based on in vitro studies, was reported at FDA hearing. John Gerber, a member of the FDA panel & HIV clinical pharmacologist, felt that there will not be clinical drug-drug interactions, despite lingering questions by one panel member.
The committee voted unanimously, 17-0, to approve the drug. In discussion, committee members supported entecavir approval for firstline and secondline therapy. They also recommended comparative studies be conducted between entecavir and adefovir & tenofovir. The FDA usually follows the recommendations of this committee. The FDA took the hearing discussions and recommendations as advise and will issue a determination.
Worldwide 350-400 million people have chronic hepatitis B infection. 15-40% develop cirrhosis, liver failure or liver cancer. About 1 million people per year die from HBV associated liver disease. In the USA about 1.25 million people have chronic hepatitis B. 10-20% of people develop chronic hepatitis B after infection. 5-8% of people with HIV also have chronic hepatitis B.
The committee reviewed results from 4 pivotol phase III studies. And considered the use of the drug for firstline and secondline treatment for chronic hepatitis B infection. Studies 022, 026, and 027 compared patients receiving entecavir or lamivudine (3TC). Study 022 was conducted in patients who were treatment naive who were hepatitis B e antigen positive. Study 027 was conducted in patients who were hepatitis B e antigen negative. Study 026 was conducted in patients who were lamivudine refractory.
The committee commented favorably on the potency, tolerability, and resistance profile of entecavir. Entecavir was compared to lamivudine in phase III studies, which were 48 weeks in length. Entecavir was found to be more potent, of equal safety & tolerability, and had a favorable resistance profile. As you may know lamivudine is considered a safe & easy to tolerate drug. The side effects from the studies are reported below in tables. After one year of therapy in studies they have not observed any resistance to entecavir. This was discussed by the committee, and it was generally thought that over time resistance to any drug develops.
Four parameters were considered when evaluating the efficacy of entecavir: the percent of study patients below 400 copies/ml, log viral load reduction from baseline to week 48, HBeAg seroconversion rate, and ALT normalization.
Under discussion today was the finding of tumors in rodents in pre-clinical studies of entecavir. The FDA cancer advisory group met & discussed this before today, and felt this finding was relevant to humans. This was the subject of much discussion at the hearing. The committee & the FDA felt that we cannot extrapolate the findings in rodents to humans. There may be a risk for humans but we cannot tell based on studies in rodents. They talked about how similar cancerous toxicities were observed preclinically with certain HIV antiretrovirals. The FDA & the committee considered the risk/benefit ratio, weighing the worldwide problem of hepatitis B & the seriousness of this disease, the need for more therapies, and the finding of tumors in rodents. Every committee member and the FDA felt that the benefit of approving entecavir outweighed potential risks. HBV can lead to liver cancer, cirrhosis, and death. All three drugs currently approved for HBV treatment have limitations. Interferon is difficult to tolerate. Adefovir has potential nephrotoxicity. And lamivudine is associated with significant drug resistance. The FDA & the committee found that entecavir should be approved for firstline and secondline HBV therapy. Due to the finding of tumors in rodents taking entecavir and the potential for humans to develop malignancies, Bristol Myers Squibb proposed a post approval safety study that would evaluate the risk for cancers developing in patients 5 to 8 years after starting entecavir, So far in the clinical studies there does not appear to be malignancies associated with entecavir. The FDA and the committee expressed satisfaction with a proposed Safety Study. The details of the study design was the subject of much discussion today, however.
Regarding efficacy of enyecavir; the percent of patients in phase 3 studies achieving <400 copies/ml (HBV DNA PCR
The data presented today showed entecavir is a potent drug against HBV. The baseline viral load in study 022 (in treatment-naive) was 9.66 logs, and the mean viral load reduction was 7 logs in study 022 for patients taking entecavir vs -5.5 log for patients receiving lamivudine. In study 027 in naives the baseline viral load was lower at 7.58 log, and the HBV DNA reductions were -5.2 log for entecavir and -4.7 log for lamivudine. These differences were significant. The side effect profiles from the studies showed entecavir to be similar to lamivudine in safety and tolerability, and entecavir was a little better in terms of the incidence of on treatment ALT flares (2% vs 4%). There appeared to be a slightly higher incidence of CNS side effects associated with entecavir compared to lamivudine. The HbeAg seroconversionb rates in study 022 were 21% for entecavir & for lamivudine 18%; this difference was not significant.
Study 026 compared entecavir to lamivudine in lamivudine refractory patients. In this study the dose of entecavir was increased from the 0.5 mg once daily dose in the studies in treatment naives to 1.0 mg once daily to overcome resistance. Patients who were lamivudine refractory switched to entecavir and they were compared to patients who remained on lamivudine. Again, the patients who switched to entecavir did better. HBV DNA <400 c/ml: 22% for entecavir vs 1% for lamivudine, the difference was significant. The baseline viral load in this study was 9.36 log, and the mean reduction in HBV DNA by PCR was -5.1 log for patients who switched, and 8% of these patients had an HBeAg seroconversion. 65% of the patients who switched to entecavir had ALT normalization, compared to 17% of patients remaining on lamivudine. In each of these parameters, entecavir outperformed lamivudine.
The primary efficacy endpoints for the Phase 3 studies were based on the change in liver histology over the initial 48 week study period when patients received blinded study drug. Histologic improvement was defined as > 2 point decrease in the Knodell necroinflammatory score with no worsening in the fibrosis score. ETV performed better for each of the individual components of the overall histologic improvement, > 2 point decrease in Knodell necroinflammatory score and no worsening in Knodell fibrosis score.
For overall histologic improvement in study 022 (treatment-naive hepatitis B e Antigen positive patients), 72% of patients receiving ETV 0.5 mg daily achieved this endpoint vs 62% of patients receiving lamivudine. This was significant (<0.03). In study 027, 70% of patients receiving entecavir & 61% receiving lamivudine had overall histologic improvement. This was also significant. No worsening in fibrosis (89% vs 82%) and necroinflammatory 2 point decrease (84% vs 79%), two key endpoints, were also better in the patients receiving entecavir. These differences were also significant. Further data from the histological efficacy can be viewed in tables below.
ETV was equivalent (non-inferior) to LVD for the secondary histologic endpoint of improvement in Ishak fibrosis score in the nucleoside-naive Studies 022 and 027 but was superior in the LVD-refractory patient population in Study 026.
There was discussion about pediatric use. BMS applied for approval of pills & oral solution. But concern was expressed by committee member that if the oral solution is approved it will be used in kids. Without PK & relevant safety data this could be problematic. On the other hand, committee members said they would like to have the oral solution for adults with renal insufficiency and for those who can't take pills.
The FDA issued this briefing document prior to the hearing.
Some of the data in this document is a little different than that presented at the hearing but not much, just a few points here & there. But the essence is the same & the statistically significant data remains significant. The FDA said their analyses were in agreement with BMS. Regarding the finding of cancer in rodents, it was reported that preclinical studies in lamivudine did not find cancers in animals. But in preclinical studies of adefovir Gilead was unable to explore high dosing because of concerns regarding nephrotoxicity. In animal testing for a new drug animals are administered doses much higher than given humans to test the limits of causing toxicities. The FDA discussed how similar findings of toxicities or I think cancers were found in certain HIV antiretrovirals, which received FDA approval.
Executive Summary: Regulatory Issues and Purpose of Meeting
This briefing document provides background information and the FDA perspective on the New Drug Applications (NDAs 21-797 and 21-798) submitted by Bristol-Myers Squibb (BMS) for entecavir (ETV), a nucleoside analogue intended for the treatment of chronic hepatitis B virus (HBV) infection in adults. The information presented in this document represents the preliminary findings and opinions of the primary reviewers from each discipline based on their review of the submitted material. The material included in this briefing document and other material presented by the applicant will be the subject of a meeting of the Antiviral Drug Products Advisory Committee to be held on March 11, 2005.
Entecavir is a guanosine nucleoside analogue with selective activity against HBV; it has no activity against other hepatitis viruses or HIV. The applicant's clinical development program was designed to assess the safety and efficacy of ETV in a variety of distinct patient populations recruited from a global network of investigators. The Advisory Committee will be asked to review and discuss issues related to the strength and completeness of the clinical database, the risk/benefit assessment of the drug in the context of non-clinical data, and the appropriateness for further post-marketing development and pharmacovigilance.
Summary of Clinical Development
The applicant has conducted an extensive global development program for ETV in the treatment of chronic HBV. At the time these studies were initiated, lamivudine (LVD) was the only approved oral treatment for chronic HBV and was chosen as the appropriate comparator for the Phase 3 studies. The preliminary opinions and discussion to follow are based on review of the 4 designated pivotal trials unless otherwise stated.
Summary of Phase 3 and Key Phase 2 Clinical Trials of Entecavir
|
|
 |
| |
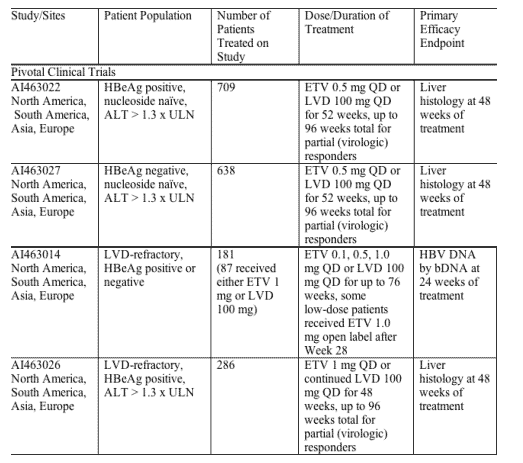
Summary of Study Designs
Study 014 was a Phase 2, randomized, double-blind, pilot study to evaluate 3 doses of ETV (0.1 mg, 0.5 mg and 1.0 mg) compared to LVD 100 mg in patients with HBV viremia while receiving LVD ("LVD-refractory"). Patients could be HBeAg-positive or negative. Patients were randomized to either continue LVD or receive ETV. Management decisions to discontinue or continue study treatment were made at Week 28 on the basis of the Week 24 assessments. Patients who achieved > 1 log reduction in HBV DNA continued blinded therapy to Week 52. Patients failing to achieve at least 1 log reduction in HBV DNA discontinued study treatment but were eligible for the rollover study (AI463901) or other available therapy. Patients remaining on study treatment were reassessed at Week 48 and management decisions were based on criteria similar to those used in other studies. Patients could continue blinded therapy for up to 76 weeks. Liver biopsy for histology was optional in this study. The primary endpoint was the proportion of patients who achieved HBV DNA levels below the LOQ by the bDNA assay (0.7 MEq/mL) at 24 weeks. This study was intended to be a dose selection study for Study 026. Only the 87 patients receiving ETV 1 mg or LVD 100 mg QD were included in the efficacy analyses and the major safety assessments presented here. The patients receiving doses less than 1 mg daily were included in only selected analyses.
The 3 pivotal Phase 3 studies utilized similar study design and endpoint analyses. Study 022 was a randomized, double-blind comparison of ETV 0.5 mg versus LVD 100 mg in HBeAg-positive, nucleoside-naive patients. Other key inclusion criteria included: age > 16 years, ALT > 1.3 x ULN, normal renal function, and compensated liver disease. Previous treatment with IFN was not an exclusion. Continuation of blinded study treatment at the end of 52 weeks was based on results of the Week 48 evaluation. Complete Responders (HBV DNA by bDNA assay < 0.7 MEq/mL and loss of HBeAg) stopped study treatment and were followed for 24 weeks off therapy to assess durability of response. Partial Responders (HBV DNA by bDNA assay < 0.7 MEq/mL but still positive for HBeAg) continued blinded therapy for up to 96 weeks or until complete response was achieved. Non-responders (HBV DNA by bDNA > 0.7 MEq/mL) discontinued study treatment but were eligible for the rollover study or other available therapy. The primary endpoint was histologic improvement on liver biopsy at 48 weeks defined as > 2 point decrease in Knodell necroinflammatory score with no worsening in fibrosis score. Secondary endpoints included improvement in Ishak fibrosis score, change in HBV DNA by bDNA assay and by PCR assay, normalization of ALT, HBeAg loss, HBeAg/HBeAb seroconversion, and various composite endpoints.
Study 027 was a randomized, double-blind comparison of ETV 0.5 mg versus LVD 100 mg in HBeAg-negative, nucleoside-naive patients. Study design was similar to Study 022 with similar management decisions occurring at Week 52. Inclusion criteria were similar to those in Study 022 except for HBeAg status. In the study treatment management algorithm, patients achieving < 0.7 MEq/mL HBV DNA by bDNA assay and ALT < 1.25 x ULN at 48 weeks were considered to have reached the composite efficacy endpoint (Composite Responders) and were eligible to discontinue study treatment and enter the follow-up phase. The primary efficacy endpoint was histologic improvement at 48 weeks defined as > 2 point decrease in Knodell necroinflammatory score with no worsening in fibrosis score. Secondary endpoints were similar except that HBeAg loss and HBeAb seroconversion were not evaluated.
Study 026 was a randomized, double-blind comparison of ETV 1 mg versus LVD 100 mg in HBeAg-positive, LVD-refractory patients with compensated liver function. Patients were randomized to either continue LVD or receive ETV. Continuation of treatment at the end of 52 weeks was based on results of Week 48 evaluation. Criteria for continuation of blinded therapy through 96 weeks were based on Complete, Partial, or Non-response and were similar to Study 022. For this study there were co-primary endpoints at Week 48: histologic improvement on liver biopsy similar to other Phase 3 studies and the proportion of patients with both undetectable HBV DNA by bDNA assay (< 0.7 MEq/mL) and normalization of ALT (defined as < 1.25 x ULN). Multiple secondary endpoints were evaluated as in the other studies.
All of the Phase 2 and 3 studies used 2 assays to measure changes in HBV DNA: HBV DNA using a branched DNA assay (Chiron/Bayer Quantiplexª v1.0) with a lower limit of quantitation (LLOQ) of 0.7 MEq/mL and HBV DNA using a PCR assay (Roche COBAS Amplicor HBV Monitorª v2.0) with an LLOQ of 300 copies/mL. Clinical management decisions were based on real-time reporting of HBV DNA by the bDNA assay results while the PCR assays were conducted at a later time. For study defined endpoint calculations, the applicant used a cut-off HBV DNA value of < 400 copies/mL for the PCR assay. Neither the bDNA assay nor the PCR assay has been approved by the FDA and both are considered investigational. However, there is considerable experience with the use of both assays in clinical trials and clinical practice.
Dose Selection
The proposed dose of ETV was selected on the basis of reductions in HBV DNA and safety and tolerability of the drug observed during short and long-term Phase 2 dose-ranging studies. In Study 004, reduction in HBV DNA over a 28-day dosing period and 28-day follow-up was greater in treatment groups receiving 0.5 and 1 mg daily than those receiving lower doses. Similarly, in Study 005, the dose of 0.5 mg resulted in greater decreases in HBV DNA over 22 weeks of dosing compared to either ETV 0.1 mg or LVD 100 mg. In these early Phase 2 studies, safety and tolerability of ETV 0.5 mg appeared to be somewhat better than ETV 1 mg, so the 0.5 mg dose was carried forward into Phase 3 trials for nucleoside-naive patients (Studies 022 and 027).
The applicant anticipated that LVD-refractory patients would require a higher dose based on in vitro data demonstrating that LVD-resistant HBV also had reduced sensitivity to ETV. Dose selection for this population was based on the interim, 24-week results from Study 014. In this study, a dose-response relationship for ETV was observed in HBV DNA reduction and the dose of ETV 1 mg was superior to 0.5 mg or to LVD 100 mg for the proportion of patients achieving HBV DNA < LLOQ by bDNA. In this study, there were no observed differences in safety and tolerability across the treatment arms. Consequently, ETV 1 mg was carried forward into the Phase 3 study in LVD-refractory patients (Study 026).
Pharmacokinetic and ADME studies of ETV document that the drug is predominately eliminated by the kidneys. Pharmacokinetics in patients with renal insufficiency, including those requiring hemodialysis or continuous ambulatory peritoneal dialysis, were studied in Study AI363011. Based on this study and pharmacokinetic modeling, the sponsor has proposed dose adjustments for patients with moderate or severe renal impairment. Elderly patients demonstrated increased ETV exposure but this could be attributed to decreased renal function and did not warrant a change in dosing based on age alone.
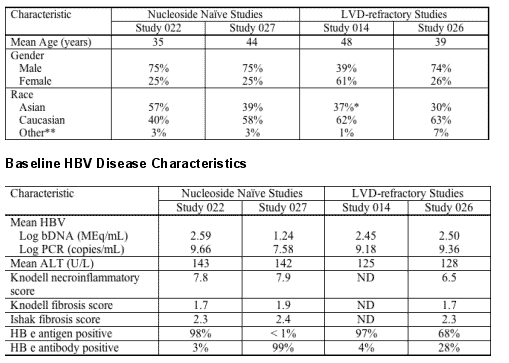
Patient Demographics
Patients who participated in the clinical trials include a representative sampling of patients with chronic HBV and compensated liver disease. Study participants were recruited from 31 countries in North America, South America, Asia, and Europe. Study demographics and baseline HBV disease characteristics for the 4 pivotal trials are summarized below (only those patients receiving ETV 1 mg or LVD were included from Study 014). For each study, demographic and baseline disease characteristics were similar across the treatment arms.
Patient Demographics
|
|
| |
 |
| |
Primary Efficacy Analysis
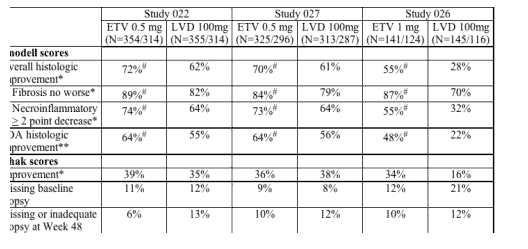
As noted above, the primary efficacy endpoints for the Phase 3 studies were based on the change in liver histology over the initial 48 week study period when patients received blinded study drug. Histologic improvement was defined as > 2 point decrease in the Knodell necroinflammatory score with no worsening in the fibrosis score. All liver biopsy specimens were evaluated by a single reader who was blinded to treatment group and biopsy order and assigned the histologic scores by both the Knodell and Ishak criteria.
The FDA statistical analysis confirmed the applicant's analysis of the primary efficacy endpoint. The applicant's analysis included patients with available baseline biopsy data and counted those with missing or inadequate Week 48 biopsy data as treatment failures. Although the studies were originally designed to show non-inferiority of ETV to LVD, this analysis demonstrated that patients receiving ETV experienced superior overall histologic improvement compared to LVD in all 3 studies (P values < 0.02 in all studies). ETV performed better for each of the individual components of the overall histologic improvement, > 2 point decrease in Knodell necroinflammatory score and no worsening in Knodell fibrosis score.
The FDA statistical reviewer also conducted sensitivity analyses of the primary histologic endpoint that included several methods of imputing missing data. A more conservative analysis included all patients who received study drug and counted patients with missing baseline or Week 48 biopsy as treatment failures. This analysis continued to show superiority of ETV compared to LVD (P values < 0.03 in all studies) in both nucleoside-naive and LVD refractory patients.
ETV was equivalent (non-inferior) to LVD for the secondary histologic endpoint of improvement in Ishak fibrosis score in the nucleoside-naive Studies 022 and 027 but was superior in the LVD-refractory patient population in Study 026. Sensitivity analyses were also conducted for the secondary histologic endpoints and supported the conclusion that ETV was no worse than LVD as measured by the Ishak score. Histologic efficacy results are summarized in Table 4 below.
Histologic Efficacy Assessments at 48 Weeks in Studies 022, 027, and 026
|
|
| |
 |
| |
N = number receiving study treatment /number with evaluable baseline liver biopsy.
*Primary endpoint: > 2 point decrease in Knodell necroinflammatory score with no worsening of Knodell fibrosis score. Individual components of primary endpoint are also shown. Efficacy calculations based on analysis of patients with available baseline biopsy, with missing Week 48 biopsy = failure.
**FDA sensitivity analysis: Efficacy calculation based on same definition of improvement but analysis of all patients who received study drug, with missing baseline or Week 48 biopsy = failure.
#ETV significantly better than LVD, P values all < 0.03.
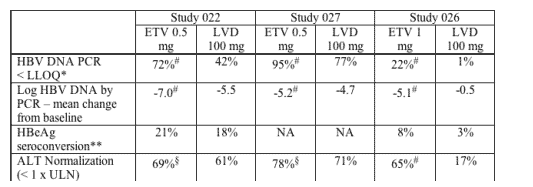
Secondary Efficacy Analysis
Treatment effects of ETV compared to LVD were also assessed for a number of secondary endpoints using virologic, serologic and biochemical measurements. Although the Phase 3 studies were originally designed using the HBV bDNA assay for treatment management decisions, discussions with the FDA prior to submitting the NDA identified the HBV PCR assay as the assay that provided better discrimination in virologic endpoints.
FDA analyses confirmed the applicant's results for the following secondary endpoints: proportion of patients with HBV DNA below LLOQ by PCR at Week 48, change from baseline in HBV DNA by PCR at Week 48, ALT normalization, and HBeAg seroconversion (loss of e antigen and gain of e antibody).
Virologic, Serologic, and Biochemical Efficacy Endpoints for Studies 022, 027, and 026
|
|
| |
 |
| |
NA: not applicable
*LLOQ: Lower limit of quantitation calculated as < 400 copies/mL.
**HBeAg seroconversion defined as loss of e antigen with gain of e antibody.
#ETV significantly better than LVD, all P values < 0.01.
ETV significantly better than LVD, P values < 0.05
Resistance
Entecavir has demonstrated activity against HBV replication in vitro with IC50 values ranging from 0.36 nM to 3.6 nM. The active intracellular moiety of ETV, ETV-triphosphate, is a competitive inhibitor of dGTP and functions as a non-obligate chain terminator. Cell culture studies have shown that viruses with the LVD resistance-associated amino acid substitutions rtM204V/I and rtL180M in the HBV polymerase display cross-resistance to ETV, having approximately 5- to 30-fold reduced susceptibility in vitro. Resistance analyses from early Phase II studies provided evidence that substitutions at positions rtT184, rtS202, and/or rtM250 are associated with ETV resistance, but developed only when LVD resistance mutations were present. The addition of substitutions at rtT184, rtS202, and/or rtM250 together with the LVD-resistance mutations, rtL180M and rtM204V, in recombinant viruses resulted in 38- to 2,000-fold reduced susceptibility to ETV in vitro.
The efficacy of ETV was examined in both nucleoside-naive and LVD-refractory patient populations. In nucleoside-naive Studies 022 (HBeAg positive subjects) and 027 (HBeAg negative subjects), 83% (541/653) of patients on 0.5 mg QD ETV treatment were suppressed with HBV DNA < 400 copies/mL as quantified by the COBAS Amplicor HBV Monitor PCR assay at week 48 compared to 59% (363/619) of patients on 100 mg QD LVD treatment. Genotypic and phenotypic analyses of paired clinical isolates obtained at study entry and Week 48 were performed to monitor baseline and emerging amino acid substitutions and to determine their impact on virologic response to ETV. In these studies, no ETV-associated resistant substitutions (T184S/A/I, S202G, M250L) emerged in any isolate on ETV therapy by 48 weeks. Two subjects experienced virologic rebound on ETV treatment but had no detectable amino acid changes emerge on treatment and no change in phenotypic susceptibility to ETV, ADV or LVD.
Studies 014 and 026 examined the efficacy of ETV 1 mg QD compared to LVD 100 mg QD in patients with LVD-refractory HBV with prior LVD experience. In these studies, LVD-resistant substitutions rtL180M and rtM204V/I were detected in > 80% of baseline isolates from both the ETV and LVD arms and these substitutions were maintained during the study, presumably because of the selective advantage in the presence of LVD and ETV. In Studies 014 and 026, 21% (36/174) of patients on ETV were suppressed to below 400 copies/mL HBV DNA by PCR assay at week 48 compared to 1% of patients on LVD.
Genotypic analyses determined that LVD-resistance substitutions L80V, L180M, M204V or I emerged in the HBV of 17% (7/42) of patients on ETV by Week 48 in Study 014. These substitutions often arose in the context of mixtures at these sites at baseline and other LVD-resistance mutations at baseline. Despite the emergence of LVD-resistance substitutions, the viral load in 4 of 7 patients was suppressed below 300 copies/mL (LLOQ) and the other 3 subjects experienced > 2 log10 reductions in viral load at the time the isolate developed the LVD-resistant mutations. ETV-associated resistance substitutions at T184 developed on 1 mg ETV therapy in 5 (12%) patients after week 48 in Study 014 and coincided with rebounds in viral load. In Study 026, substitutions at RT residues rtI169, rtT184, rtS202 and/or rtM250 emerged on therapy in 9% (12/134) of ETV subjects with Week 48 data. In all cases, the ETV-resistant substitutions emerged when pre-existing LVD-resistant changes were present.
The supportive Study 015 examined the antiviral activity of open label ETV 1 mg QD in 9 orthotopic liver transplant (OLT) recipients who were > 100 days post-transplant and had recurrent HBV infection despite prophylaxis with anti-HBV agents. In this study, virologic rebound occurred in 6 out of 8 patients - one in the first year therapy, one in the second year, and four in the third year while 2 patients maintained HBV DNA suppression with no rebound out to 127 and 131 weeks of therapy. Seven of the eight patients showed the development of ETV-resistance substitutions at S202G or I (n=5), T184S/I/A/L/F (n=4) or M250V (n=1), and these substitutions were linked to LVD-resistant changes L180M and M204V.
The substitutions at rtI169, rtT184, rtS202 and/or rtM250 were associated with phenotypic ETV resistance. The median fold change from reference of ETV susceptibility was 38 (range 12-2139) for the ETV failure isolates (> 400 copies/mL HBV DNA) that developed ETV-resistance substitutions at 48 weeks in Study 026 (n = 15) and 83 (range 12-10022) for all ETV failure isolates from Studies 026 and 015 > 48 weeks (n = 22).
Overall, the following conclusions can be summarized from the resistance data collected in Studies 022, 027, 014, 026, and 015.
* Greater proportions of nucleoside-naive subjects (83%) with chronic HBV infection achieved HBV DNA levels < 400 copies/mL on ETV treatment compared to LVD-refractory subjects (21%).
* Genotypic or phenotypic evidence of resistance to ETV in nucleoside-naive patients chronically infected with HBV (n = 430) has not been observed up to 48 weeks of 0.5 mg QD ETV treatment in Studies 022 and 027, including 2 subjects in Study 022 who experienced a confirmed virologic rebound.
-7.4% (14/190) of LVD-refractory subjects treated with 1 mg ETV had evidence of emerging ETV-resistance substitutions by week 48.
-ETV-associated resistance substitutions at rtI169, rtT184, rtS202, and/or rtM250 emerged concomitant with LVDÐresistant mutations at rtL180 and/or rtM204 and were associated with virologic rebound upon prolonged ETV therapy.
-Overall, 4 ETV treated subjects exhibited a confirmed rebound in their HBV DNA levels of > 1 log10 by week 48:
2 isolates from Study 022 with no evidence of ETV-resistant substitutions or other genotypic changes
One isolate from Study 015 that developed an rtT184A
One isolate from Study 026 that developed an rtT184A/S
-LVD-resistance substitutions L80V, L180M, M204V or I can emerge in the HBV of patients on 1 mg ETV by Week 48. These substitutions often arise in the context of mixtures at these sites at baseline and other LVD-resistant mutations at baseline.
-Even when LVD-resistant mutations emerged in HBV on ETV therapy, ETV can suppress HBV DNA levels to below detection limits.
-> 2 log10 reductions in viral load and viral load suppression below 400 copies/mL HBV DNA can occur in subjects with LVD-resistance in their HBV at baseline when treated with 1 mg ETV.
-Cross-resistance to ETV was not observed with ADV-resistant HBV.
-HBV developing ETV resistance-associated substitutions in addition to LVD-associated resistance substitutions remained resistant to LVD.
Studies evaluating treatment responses to ETV and monitoring resistance to ETV beyond 48 weeks of dosing are ongoing.
Summary of Non-clinical Toxicology and Carcinogenicity
Overview of General Non-clinical Findings
Entecavir is efficiently phosphorylated to ETV-triphosphate (TP) by cellular nucleoside kinases. By competing directly with the natural deoxyguanosine triphosphate (dGTP), ETV-TP potently inhibits each of the 3 distinct activities of the HBV viral polymerase: priming, reverse transcription of first-strand DNA synthesis, and the DNA-dependent DNA polymerase activity responsible for second-strand DNA synthesis. Entecavir has little activity against the DNA polymerase of mitochondria.
The pharmacokinetic (PK) characteristics of entecavir in mice, rats, rabbits, dogs, and monkeys are comparable to those in humans indicating the acceptability of these species for the toxicological assessment of ETV.
Species-specific, reversible CNS inflammation was seen in dogs administered doses that achieve ~51 times the exposure to ETV in humans at clinically proposed doses. It was concluded that this is not relevant to human safety. Other target organs in repeat-dose studies in animals were the kidneys, liver, lungs, skeletal muscle and testis. Data from a 1-year study in monkeys indicated that there was no target organ toxicity in monkeys at exposures to ETV ~136 times those in humans.
Long-term dosing of ETV was evaluated in a woodchuck model of chronic HBV. In this study, woodchucks received a daily dose of ETV equivalent to the 1 mg human dose for 2 months and then were maintained with weekly dosing for up to 3 years. Viral suppression was maintained through 3 years of treatment with no evidence of emergence of resistant HBV. The applicant reported survival rates of 40% and 80% for animals treated for 14 and 36 months, respectively, compared to a survival of 4% in historical controls. Of most interest, the occurrence of hepatocellular carcinoma was significantly reduced in the animals treated long-term compared to historical control animals.
Overview of Carcinogenicity Studies
In a battery of genetic toxicology studies, ETV was an in vitro mutagen in mouse lymphocytes and clastogenic in vitro in human lymphocytes (without metabolic activation). However, ETV was negative in an Ames assay as well as a mammalian-cell gene mutation assay and a cell transformation assay. It was also negative in two in vivo assays, one for the induction of micronuclei and one for the induction of unscheduled DNA synthesis in primary liver cells.
Carcinogenicity studies in Sprague Dawley rats and CD-1 mice were conducted. Increased incidences of tumors were observed in both the studies. The results of these studies were presented to the Executive Carcinogenicity Assessment Committee (ECAC) on June 17, 2003. The key results of these studies are presented in tabular format in Appendix B. The outcomes of the two studies were as follows:
Rat Carcinogenicity Study: The oncogenicity potential of ETV was investigated in male rats at oral gavage dosages of 0.003 (low), 0.02 (mid), 0.2 (high) or 1.4 mg/kg/day (highest) and in females at dose levels of 0.01 (low), 0.06 (mid), 0.4 (high) or 2.6 mg/kg/day (highest) in comparison with untreated controls for a period of 104 weeks.
The no observed effect level (NOEL) for neoplasia was 0.2 mg/kg/day for males and 0.06 mg/kg/day for females. At tumorigenic doses, systemic exposures were 35- and 4-times that in humans (1 mg daily dose) in male and female rats, respectively.
Treatment-Associated Tumors:
1. Hepatocellular adenomas in female rats were significant (p=0.005) at the highest dose level. Combined adenomas and carcinomas in the female rats were also significant (p=0.005) at the highest dose. In female rats, the combined incidence of adenomas and carcinomas was 1% (controls), 4% (low), 5% (mid), 2% (high) and 18% (highest).
2. Brain gliomas were significant (p=0.025) at the highest dose in both male and female rats. In male rats, the incidence was 0% (controls), 2% (low), 2% (mid), 3% (high) and 7% (highest). In female rats, the incidence was 0% (controls), 0% (low), 2% (mid), 0% (high) and 5% (highest).
3. The skin fibromas in female rats were significant (p=0.025) at the high and highest doses. In female rats, the incidence was 0% (controls), 0% (low), 2% (mid), 3% (high) and 5% (highest).
Mouse Carcinogenicity Study: The oncogenicity potential of ETV was investigated in mice at oral gavage dosages of 0.004 (low), 0.04 (mid), 0.4 (high) or 4.0 mg/kg/day (highest) in comparison with untreated controls for a period of 104 weeks.
The NOEL for neoplasia was 0.004 mg/kg/day for males, based on pulmonary adenomas; for all other tumors in males and females, the NOEL was 0.4 mg/kg/day. At the tumorigenic dose in male mice, systemic exposure was 3-times that in humans (1 mg daily dose).
Treatment-Associated Tumors:
1. Lung adenomas were significant (p=0.005) in male mice (mid, high and highest) and in the female mice at the highest dose (p=0.005); lung carcinomas in both male and female mice were significant (p=0.005) at the highest dose. Combined lung adenomas and carcinomas were significant (p=0.005) in male mice at the mid, high and highest dose levels and in the female at the highest dose level (p=0.005. In male mice, the combined incidence of adenomas and carcinomas was 12% (controls), 20% (low), 26% (mid), 40% (high) and 58% (highest). In female mice, the combined incidence of adenomas and carcinomas was 20% (controls), 13% (low), 10% (mid), 35% (high) and 52% (highest).
2. Hepatocellular carcinomas in male mice were significant (p=0.005) at the highest dose level. Combined liver adenomas and carcinomas were also significant (p=0.005) at the highest dose level in the male mice. In male mice, the combined incidence of adenomas and carcinomas was 11% (controls), 9% (low), 8% (mid), 16% (high) and 25% (highest).
3. Vascular tumors in female mice (hemangiomas of ovaries and uterus and hemangiomas/hemangiosarcomas of spleen) were significant (p=0.005) at the highest dose level. In female mice, the incidence of vascular tumors was 16% (controls), 23% (low), 29% (mid), 26% (high) and 64% (highest).
The ECAC found that the carcinogenicity studies in mice and rats were adequately designed and conducted. The committee judged the results of ETV carcinogenicity studies. They concluded that ETV was a carcinogen in rodents. The committee concluded that ETV produced tumors in both species and both genders, and these results suggest a potential cancer hazard to patients.
At the request of the sponsor, the results of the carcinogenicity studies were presented to the full FDA CAC (CAC), a committee that has been designated as the arbiter of disputes between applicants and review divisions regarding the relevance of results in carcinogenicity studies. The CAC met with the applicant and the Review Team on January 7, 2005 and concluded that hepatocellular adenomas and carcinomas in female rats, skin fibromas in female rats and brain gliomas in both male and female rats were relevant. The committee also agreed that in the mouse carcinogenicity study, liver tumors in males and vascular tumors in females as well as lung tumors in both sexes were relevant to human safety evaluation.
Common Adverse Events, Serious Adverse Events, and Laboratory Abnormalities
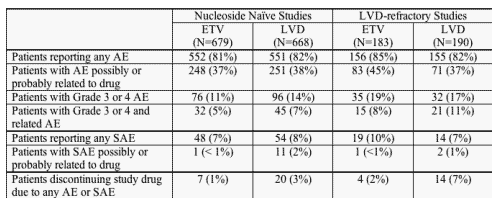
FDA review of the safety database for the pivotal trials confirmed the applicant's analysis of common adverse events (AEs), serious adverse events (SAEs), and laboratory abnormalities. All patients who received at least one dose of blinded study medication in the pivotal trials were included in the safety analyses. The review included assessment of proportions of patients who experienced AEs and SAEs according to severity, relationship to blinded study drug, and action required to manage the event (interruption or discontinuation of study drug). The safety review evaluated clinical events and laboratory abnormalities according to assigned treatment and over 2 study periods (on blinded treatment and off treatment). Organ systems identified as potential targets in the non-clinical animal studies were specifically reviewed. The review evaluated safety in each of the studies individually and also pooled the analyses of nucleoside-naive patients (Studies 022 and 027) and LVD-refractory patients (Study 014 groups receiving ETV 1 mg or LVD and Study 026). Summary results of the pooled analysis will be presented below. Minor differences between the applicant's results and the FDA's results can be attributed to slightly different methods of defining visit windows and conducting the analyses and do not alter the final conclusions.
AEs were reported frequently in the nucleoside-naive patients although there were few differences in the pattern of AEs reported by ETV-treated patients compared to LVD-treated patients. On treatment AEs reported in > 5% of patients in either arm in the nucleoside-naive studies included: headache, upper respiratory infection, nasopharyngitis, cough, pyrexia, abdominal pain, diarrhea, fatigue, arthralgia, dizziness, nausea, influenza, sore throat, rhinorrhea, dyspepsia, increased ALT, increased blood amylase, back pain, and myalgia. Most of the reported events were mild and not considered related to study treatment. The proportions of patients with reported AEs considered by the investigators to be possibly or probably related to blinded study drug were similar in the 2 treatment groups (ETV 37%, LVD 38%). AEs were reported in smaller numbers of nucleoside-naive patients during the off-treatment follow-up period and very few events were observed in > 5% of patients (increased ALT in ETV 3% and LVD 11%, headache in ETV 5% and LVD 6%). During the off-treatment period, only increased ALT occurred more frequently in LVD patients than ETV patients. Other AEs occurred in similar numbers of patients in both groups.
The pattern of commonly reported AEs was very similar in the LVD-refractory patients. On treatment AEs reported in > 5% of patients in either arm in the LVD-refractory studies included: headache, upper respiratory infection, abdominal pain, fatigue, cough, nasopharyngitis, pyrexia, diarrhea, arthralgia, dizziness, nausea, sore throat, dyspepsia, ALT increased, back pain, and myalgia. Most of the events were described as mild and not related to study drug. In this population, increased ALT was reported more frequently in patients receiving LVD (11%) than in those receiving ETV (3%) and fever and sore throat were reported more frequently in ETV patients (9% and 7%, respectively) than in LVD patients (4% and 2%). Reflective of the relatively small proportion of LVD-refractory patients who entered off-treatment follow-up, few patients experienced AEs during the off-treatment period. There were no significant differences in the pattern of off-treatment AEs between the treatment groups.
The number of patients who developed SAEs (death, hospitalization, cancer, congenital anomaly, life-threatening condition, or other medically significant event) while on study was small. Similarly, the number of patients discontinuing their assigned study drug because of an AE or SAE was low, 1% for ETV-treated patients and 4% for LVD-treated patients.
Proportions of Patients Reporting Adverse Events or Serious Adverse Events while on Study Drug
|
|
| |
 |
| |
Clinical laboratory monitoring for safety included assessments of routine hematology and coagulation studies, serum biochemical studies, and urinalysis at each study visit. Confirmatory review of the applicant's analysis consisted of evaluation of the numbers of patients who exhibited extreme laboratory values (Grade 3 or 4). As with the clinical AEs, laboratory abnormalities were compared across treatment groups and for both on-treatment and off-treatment periods. Results are presented for pooled nucleoside-naive patients and pooled LVD-refractory patients during the on-treatment period.
The most commonly observed hematologic/coagulation abnormalities in the nucleoside-naive patients were prolonged PT and increased INR. During the on-treatment period, prolonged PT was identified in 36% of ETV patients and 32% of LVD patients. Increased INR was observed in 28% of ETV patients and 24% of LVD patients. However, Grade 3 or 4 abnormalities of PT and INR were observed in 2% and 1%, respectively, of ETV patients and < 1% each of LVD patients. Abnormalities in other hematologic parameters were rare and balanced across treatment groups. Among LVD-refractory patients, PT and INR abnormalities were the most commonly observed abnormalities (ETV 34% and 32%, respectively, LVD 36% and 38%). In this population, 4% of ETV patients compared to 11% of LVD patients had low platelet counts at some time on-treatment but Grade 3 or 4 hematologic abnormalities were rare.
There were few significant abnormalities in serum biochemical tests identified in either the nucleoside-naive or LVD-refractory patient populations. Elevations of pancreatic enzymes, increased creatinine, and abnormalities in electrolytes occurred rarely and with similar prevalence across the treatment groups. The most commonly observed biochemical abnormalities were elevations in liver transaminases. Since increased ALT was one of the screening criteria for all clinical trials, most patients had documented transaminase abnormalities at baseline. In general, mean ALT and AST levels decreased among nucleoside-naive patients on treatment in both treatment arms. For example, among ETV patients with ALT values in the nucleoside naive safety dataset, mean ALT decreased from 141 U/L at baseline to 36 U/L at Week 48. Among LVD patients in the same population, mean ALT decreased from 145 U/L at baseline to 45 U/L at Week 48. In this population, significant numbers of patients experienced Grade 3 or 4 ALT elevations after the baseline value: 21% of ETV patients and 25% of LVD patients. Among LVD-refractory patients, Grade 3 or 4 ALT elevations were also reported in a significant number of patients and were more common in the LVD group: 19% of ETV patients and 31% of LVD patients. Additional information regarding the occurrence of ALT "flares" will be presented in more detail in Section 5.4.1.
Deaths
There were a total of 15 deaths during treatment with study drugs during all of the reported ETV trials. Across the pivotal trials, 12 deaths were balanced between patients receiving ETV and those receiving LVD (see Table 8). In the nucleoside-naive studies there were 6 deaths among the 1347 subjects (0.4%) while in the LVD-refractory studies there were 6 deaths in the 373 (2%) patients receiving study drug. Three additional deaths were reported from the Phase 2 supportive studies. None of the deaths were considered by the investigators to be related to study drugs but one death was thought to be possibly related to withdrawal of study drug and resulting hepatic decompensation.
ALT "Flares" and Hepatic Adverse Events
Acute exacerbations of hepatitis, sometimes called "flares," represent an important safety issue in the treatment of chronic HBV infection. Flares have been described during treatment with all of the approved drugs and after discontinuation of drugs that have activity against HBV. During the ETV development program, the applicant tracked hepatitis flares using a standardized definition, the occurrence of ALT value at least twice the baseline value and 10 x the ULN. Safety data was also reviewed to evaluate the occurrence of these ALT flares in combination with other clinical hepatic events or other laboratory abnormalities consistent with worsening liver function. During the clinical trials, ALT flares were separated into those occurring during treatment and those occurring after discontinuation of study drugs.
ALT flares occurred infrequently in nucleoside-naive patients during the on-treatment period: 15 ETV patients (2%) and 27 LVD patients (4%). Flares appeared clustered in the first 12 weeks on study drugs and again in the later stages of the on-study period. The flares occurring during the first 12 weeks on study were often accompanied by decreases in HBV DNA and did not necessitate discontinuing study drug (9 ETV patients and 11 LVD patients). Flares occurring later in treatment (2 ETV and 12 LVD) were often accompanied by increases in HBV DNA and more often prompted study drug discontinuation. One LVD patient had both an early and a late flare, the second prompting drug discontinuation. Among the nucleoside-naive patients, 1 ETV patient and 3 LVD patients discontinued study drug due to flares and one LVD patient developed hepatic decompensation with hepatorenal syndrome and died. During the off-treatment follow-up, 15/414 (4%) ETV patients and 30/377 (8%) LVD patients with off-treatment data experienced ALT flares.
ALT flares were documented more often among patients in the LVD-refractory trials (including Study 026 patients and the Study 014 patients who received ETV 1 mg or LVD). In this population, 4 ETV patients (2%) and 19 LVD patients (10%) experienced ALT flares while receiving study drug. Six LVD patients discontinued study drug because of ALT flares. During the off-treatment follow-up, 4/56 ETV patients (7%) and 4/31 LVD patients (13%) with follow-up data experienced ALT flares.
Clinical AEs related to the hepatobiliary system or to hepatic (laboratory) investigations reported as AEs were also tabulated. Among the nucleoside-naive patients, 57 ETV patients (8%) and 87 LVD (13%) patients reported a clinical or laboratory AE related to the hepatobiliary system while receiving study treatment. Most of these events represented increases in ALT, AST, or bilirubin. The most common clinical AE reported was "hepatic pain" reported in five patients in each treatment group. A total of 13 nucleoside-naive patients experienced non-malignant hepatobiliary SAEs while on study treatment: 3 ETV patients and 10 LVD patients. These events included: increased ALT, portal vein thrombosis, and cholelithiasis in ETV patients and increased ALT, AST, and/or bilirubin (7), cholecystitis (2), and hepatic failure in LVD patients. Hepatic malignancies were considered SAEs but will be discussed separately.
Among LVD-refractory patients, 22 ETV patients (12%) and 32 LVD patients (17%) experienced a hepatobiliary clinical or laboratory AE during the treatment period. The most commonly reported events were clinically significant abnormalities of ALT, AST, and bilirubin. The most common clinical AEs reported were cholelithiasis (1 ETV patient and 2 LVD patients) and "liver lesion" (2 ETV patients). Four patients experienced non-malignant, hepatobiliary SAEs while on study treatment: acute cholecystitis/ cholelithiasis and severe hepatitis in ETV patients and cholecystitis and hepatic flare in LVD patients.
Malignancies
Evaluation of malignancies was of special interest during the review process because chronic HBV is known to be a strong risk factor for development of hepatocellular carcinoma (HCC) and because the results of the rodent carcinogenicity studies suggested that ETV might itself be a potential carcinogen. Early pre-clinical studies using a woodchuck model suggested that administration of ETV to HBV-infected woodchucks decreased the occurrence of HCC in animals that were maintained on the drug long-term.
The applicant's initial evaluation of malignancies for the NDA included data from 10 Phase 2 and 3 studies through a cut-off date of May 28, 2004. This analysis includes data from 1392 patients initially treated with ETV, 899 patients treated with LVD, and 108 patients who initially received placebo. Of the patients initially randomized to receive placebo, 105 subsequently received ETV and are included with that group for a total of 1497 patients receiving ETV.
As of the cut-off date reported in the NDA, a total of 27 malignancies had been identified in 26 patients (17 ETV patients and 9 LVD patients). No malignancies were diagnosed among the 108 patients who originally received placebo in early clinical trials. In addition, there were 5 patients (3 ETV and 2 LVD) who were reported to have lesions that were categorized as pre-malignant or unclassifiable. These patients and their diagnoses are summarized in Appendix C.
The Medical Officers reviewed all narrative summaries and Case Report Forms of patients with reported malignancies and it was concluded that none of the case descriptions were unusual or reported the occurrence of rare tumor types. Some of the reported malignancies occurred in patients who were relatively young for a tumor type but not outside the reported range (eg., breast cancer in a 30 year old woman, HCC in a 26 year old man). Some malignancies were identified after a relatively brief exposure to ETV or LVD, suggesting that study drug use had little impact on the development of the cancer in those cases. Others were identified after the patient received study drug for over a year. Even this is a relatively short reporting period for assessing carcinogenic potential.
Little information is available regarding prior medical history or risk factors for malignancy for patients enrolled in the ETV clinical trials. Six of the patients reported to have malignancies were known to have had previous malignancies. Patient AI463015-16-2010 had a history of HCC prior to transplant and then was diagnosed with renal cancer 779 days after beginning ETV. Patient AI463022-115-10657 had a history of nephrectomy for renal cell carcinoma prior to study and then developed multiple metastatic lesions in the brain, bones, and lungs (no biopsy diagnosis) after 358 days on LVD. Patient AI463022-80-10451 had a history of gastric cancer prior to study enrollment and developed recurrence of her gastric cancer and metastases after 277 days on LVD. Additionally, 3 patients who reported basal cell or squamous cell carcinoma of the skin during study observation had a previous history of skin cancer.
The applicant calculated the rate of malignancies over time for patients receiving ETV or LVD in the clinical trials. They note that the overall rate of malignant neoplasms was 8.5 per 1000 patient years of observation for patients receiving ETV and 7.8 per 1000 patient years for patients receiving LVD. This compares to rates of 9.7 per 1000 patient years for all cancers in patients with chronic HBV and 3.8 per 1000 patients years in patients without evidence of HBV calculated from a U.S. cohort study commissioned by BMS. For HCC, the most commonly reported malignancy, the rate was 3.5 per 1000 patient years for ETV patients and 3.4 per 1000 patient years for LVD patients. These rates compare to rates of 4.6 per 1000 patient years in patients with chronic HBV and 0.02 per 1000 patient years in the non-HBV comparator group calculated from the U.S. cohort study.
Addition of new cases reported in a recent Safety Update containing data through August 17, 2004, brings the total number of patients with identified malignancies in the ETV development program to 37. Of these patients 28 were in the randomized populations: 19/1497 ETV patients (1.3%) and 9/899 LVD patients (1%). Nine patients were in special study populations not previously analyzed (decompensated, HIV/HBV co-infected, or receiving dual therapy): 3 receiving ETV alone, 2 receiving adefovir (ADV) alone, and 4 receiving combination therapy with ETV+LVD. There was less information available on these patients for review but summary data is included in the table in Appendix A.
Summary of Entecavir Clinical Pharmacology
The clinical pharmacology and pharmacokinetic profile of ETV have been defined in healthy subjects and HBV-infected patients. These studies show ETV demonstrates the following clinical pharmacology characteristics:
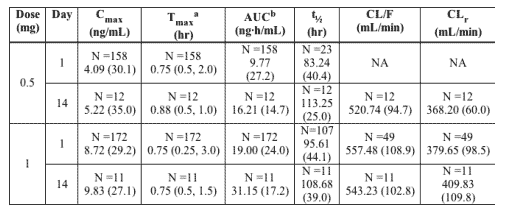
-An integrated summary of ETV single and multiple dose pharmacokinetic parameters following administration of the proposed therapeutic doses (0.5 mg and 1 mg) in the fasted state are presented in the following table.
|
|
| |
 |
| |
Data presented as geometric mean (CV%) unless otherwise specified.
NA Not available
a Data presented as median (minimum, maximum).
b AUC is AUC(0-T) on Day 1 and AUC(TAU) on Days 7 & 14
-ETV plasma concentrations are similar following administration of the tablet or oral solution.
-Following the administration of ETV at the clinically relevant doses of 0.5 or 1 mg, the systemic exposure demonstrated approximately 2-fold accumulation. ETV has an apparent terminal half life of approximately 130 hours and an effective half life for accumulation of approximately 24 hours. Trough concentrations indicated that steady state was attained by approximately 9 to 10 days following once-daily dosing.
-ETV exposure decreased by approximately 20 % following a high-fat or light meal compared to fasted conditions. The proposed label recommends ETV be administered on an empty stomach (at least 2 hours before and at least 2 hours after a meal).
-The protein binding of ETV in human serum is low (approximately 13%), and ETV uniformly distributes between plasma and red blood cells in whole human blood.
-Following administration of a 1 mg dose of [14C]-ETV, 75% of the total radioactivity (TRA) administered was recovered in the urine and 6% was recovered in the feces. Approximately 70% of the administered ETV dose was excreted as unchanged drug in urine over 14 days of collection, suggesting an estimated bioavailability ³ 70%.
-Renal excretion of unchanged drug is the primary route of ETV elimination, while biliary excretion plays a minor role. Values for renal clearance of ETV were greater than the glomerular filtration rate, indicating that the excretion of ETV by the kidneys occurs via a combination of glomerular filtration and net tubular secretion.
-ETV is not a substrate for P-glycoprotein.
-Several in vitro studies indicate that ETV is not a substrate, inhibitor, or inducer of the CYP450 enzyme system. The only metabolites detected in human plasma, urine, and feces were minor amounts of phase 2 metabolites, namely, glucuronide and sulfate conjugates. No oxidative metabolites of ETV were detected indicating that CYP450 does not play a role in the metabolic clearance of ETV in vivo.
-There were no significant pharmacokinetic interactions between ETV and LVD, ADV, or tenofovir in Phase 1 drug interaction studies. In addition, an in vitro study showed that co-administration of stavudine, didanosine, abacavir, zidovudine, LVD, or tenofovir with ETV had no effect on anti-HBV and/or anti-HIV-1 activity of any of the compounds.
-In subjects with selected degrees of renal impairment, as renal function declined mean apparent total body clearance and renal clearance of ETV decreased. This decrease in clearance resulted in a longer half life and greater exposure to ETV, as compared to subjects with normal renal function. Additionally, hemodialysis removed about 13% of the ETV dose, while continuous ambulatory peritoneal dialysis (CAPD) removed < 1% over 7 days in subjects with severe renal impairment. Based on these findings, dosage reduction of ETV is warranted in the presence of renal impairment. Modeling and simulation of multiple-dose administration of the proposed dosage recommendations in patients with varying degrees of renal function was performed. Based on the safety margin defined by the Phase 1 program, a target range of exposure was defined as two times the geometric mean steady state AUC value in subjects with normal renal function (maximum) and the lowest predicted value for subjects with normal renal function (minimum). Specific dosage recommendations for patients with renal impairment are currently under review.
-Hepatic impairment had a negligible impact on ETV exposure, and no dose modification based on the presence of hepatic impairment is necessary.
-Differences in ETV PK between Asian and non-Asian populations were observed. Cmax and AUC following multiple 0.5 mg dosing of ETV were approximately 50% and 20% higher in healthy Asian subjects versus healthy non-Asian subjects. Weight-normalized CL/F values were comparable between the Japanese and non-Asian study populations, suggesting the ethnic differences in exposure between Asian and non-Asian populations may be attributable to differences in body weight, but small sample sizes across these study populations preclude definition of an effect of race on ETV pharmacokinetics.
-ETV exposure was approximately 29% higher in elderly compared to young subjects, a disparity attributable to differences in renal function.
-No significant gender-related differences in ETV pharmacokinetics were observed.
- differences in ETV exposure were observed between healthy subjects and in HBV infected subjects. In comparison to healthy subjects, ETV AUC was about 30% & 71% higher after multiple daily dosing of 0.5 & 1 mg, respectively. In HBV subjects post-orthotopic liver transplant (OLT), mean Cmax was increased by about 42% and the mean AUC was increased by about 116% compared to healthy subjects following 14 days of oral 1 mg ETV. This increase in Cmax & AUC in OLT patients was consistent with the degree of renail impairment in these subjects.
-No dose- or concentration-dependent relationships between QT interval (with Bazett's or Fridericia's correction) or change in QTc were observed following ETV doses up to 20 mg for up to 14 days or as a single dose of 40 mg in healthy volunteers. In contrast, a slight concentration-dependent effect on PR interval was observed following ETV doses of up to 20 mg for 14 days (slope = 0.124). These findings were based on a retrospective analysis of 5 controlled Phase 1 studies. The slight prolongation in PR in this retrospective analysis is not expected to be clinically significant.
Issues for Committee Discussion
Risk/benefit Assessment
Several issues must be considered in determining the overall risk/benefit of ETV in the treatment of chronic HBV and how it might fit into the current treatment armamentarium. Chronic HBV remains a major contributor to the global rates of cirrhosis, HCC, and mortality. ETV achieves reliable drug exposure in patients, has few significant drug-drug interactions, and dosing can be reasonably adjusted in patients with impaired renal function using the oral solution formulation. We conclude from the pivotal clinical trials and supportive studies that ETV effectively reduces the HBV viral burden and leads to improvement in liver histology and normalization of liver transaminases in patients receiving the drug for 48 weeks. It achieved these endpoints in a greater proportion of patients than did LVD in both nucleoside-naive patients and LVD-refractory patients. The general tolerability and safety profile of ETV was similar to that of LVD over the observed dosing and post-dosing periods. Assessment of the drug in dosing beyond 48 weeks is ongoing.
These positive findings from the ETV studies must be weighed against findings that are less clearly understood. While resistance to ETV has not yet emerged in nucleoside-naive patients during the observation period, patients with prior resistance to LVD appear to be at risk for development of reduced susceptibility to ETV. This may have a significant impact on long-term dosing as the clinical trials continue.
Even more uncertainty emerges in the assessment of the potential risk that ETV may be a carcinogen given the results of the rodent studies. This issue is complicated by the oncogenic properties of HBV itself and by accumulating animal and human data suggesting that HBV treatment may prevent or delay the occurrence of HCC. LVD has been studied in similar carcinogenicity studies and has been found to have no carcinogenic effects even at high doses. Carcinogenicity studies with ADV were limited by an inability to deliver high doses of the drug to rodents because of significant renal toxicity. It is possible that the dose-related pulmonary tumors identified in mice receiving ETV are species specific. However, multiple other tumors were identified in both mice and rats receiving high doses of ETV, raising the possibility that the drug may have broader carcinogenic effects. It is always difficult to extrapolate animal carcinogenicity data to human risk and so we are unable to determine the magnitude of the risk to humans from currently available data. To our knowledge, there has not been a public discussion of an acceptable level of carcinogenicity risk in relationship to chronic HBV treatment.
Issues for discussion:
The risk/benefit assessment of ETV in the context of the available clinical safety and efficacy data and non-clinical carcinogenicity data.
Pediatric Development
Although HBV vaccination has been universally recommended for infants in the U.S. and many other countries, there remains a substantial population of pediatric patients affected by chronic HBV. These patients are at high risk for development of HCC over their lifetimes. At present, interferon-_ and LVD are approved for treatment of chronic HBV in pediatric patients. The Review Team asked BMS to delay its pediatric development program after the results of the rodent carcinogenicity studies were reported. We proposed that pediatric drug development could proceed if a full assessment of the potential risks and benefits of the drug in the adult population determined that ETV might provide significant benefit for pediatric patients.
Issues for discussion:
1. Potential risks and benefits of proceeding with development of ETV for the treatment of chronic HBV in pediatric patients.
2. Additional information needed about either ETV or the pediatric population in order to proceed.
Post-marketing Commitments
The applicant has proposed a post-marketing pharmacovigilance plan with the objectives of providing longer-term safety data. The events of most interest include: death, malignancy (HCC and other), hepatitis flares occurring on-treatment and post-dosing, and need for liver transplantation. Currently enrolling rollover studies will be continued with Studies 050 and 901 extending dosing for up to 3 years and Study 049 extending post-treatment observation for up to 5 years. These studies will capture many of the patients completing other Phase 2 and 3 studies. The applicant will also provide periodic safety updates summarizing AEs and SAEs of special interest from both the on-going clinical development program and from spontaneous post-marketing reporting.
In order to provide additional assurance that ETV does not pose an increased risk of malignancy, the applicant has proposed conducting a randomized, prospective, post-marketing observational study. The protocol synopsis proposes to enroll approximately 12,500 patients with chronic HBV who are eligible for treatment and randomize them to receive either ETV or other standard nucleoside/nucleotide therapy. This large, simple study will track overall mortality, liver transplantation, and malignancy (HCC and non-HCC) for 5 years after the last patient is enrolled. We have encouraged the applicant to further develop a full protocol for review but recognize that there are inherent limitations in a study of this type. In spite of its length, the study may not be able to identify a cancer risk with a long latency and, other than HCC, no specific tumor type can be targeted for surveillance. Different proportions of patients from specific geographic areas or at different stages of disease may not be balanced across the treatment arms and patients may change treatments during the observation period increasing the difficulty interpreting results. However, we accept that the applicant's proposal represents a good faith effort to identify 5 to 8-year cancer risk.
Issues for discussion:
Appropriateness of the applicant's proposed pharmacovigilance plan to address clinically relevant issues.
|
|
| |
| |
|
|
|