Michael Stumvoll, Barry J Goldstein, Timon W van Haeften
Lancet, April 9 2005; 365: 1333-46
Third Medical Department, University of Leipzig, Leipzig, Germany (Prof M Stumvoll MD); Division of Endocrinology, Diabetes and Metabolic Diseases, Department of Medicine, Jefferson Medical College of Thomas Jefferson University, Philadelphia, PA, USA (Prof B J Goldstein MD); and Department of Internal Medicine G 02·228, University Medical Centre Utrecht, PO Box 85500, NL 3508 GA Utrecht, Netherlands (T W van Haeften MD)
Type 2 diabetes mellitus has become an epidemic, and virtually no physician is without patients who have the disease. Whereas insulin insensitivity is an early phenomenon partly related to obesity, pancreas ß-cell function declines gradually over time already before the onset of clinical hyperglycaemia. Several mechanisms have been proposed, including increased non-esterified fatty acids, inflammatory cytokines, adipokines, and mitochondrial dysfunction for insulin resistance, and glucotoxicity, lipotoxicity, and amyloid formation for ß-cell dysfunction. Moreover, the disease has a strong genetic component, but only a handful of genes have been identified so far: genes for calpain 10, potassium inward-rectifier 6·2, peroxisome proliferator-activated receptor gamma, insulin receptor substrate-1, and others. Management includes not only diet and exercise, but also combinations of anti-hyperglycaemic drug treatment with lipid-lowering, antihypertensive, and anti platelet therapy.
CONTENTS
Diagnosis
Heredity in type 2 diabetes mellitus
Pathophysiology of hyperglycaemia
Insulin resistance
ß-cell dysfunction
Genetic factors
Management of hyperglycaemia
Management of other cardiovascular risk factors
Diabetes prevention
Future aspects
Diabetes mellitus has reached epidemic proportions and affects more than 170 million individuals worldwide. Global estimates for the year 2010 predict a further growth of almost 50%, with the greatest increases in the developing countries of Africa, Asia, and South America.1 In more developed societies, the prevalence of diabetes mellitus has reached about 6%,2 and, even more alarmingly, among obese white adolescents 4% had diabetes and 25% had abnormal glucose tolerance.3 Some 90% of diabetic individuals have type 2 (non-insulin-dependent) diabetes mellitus, and within this category no more than 10% can be accounted for by monogenic forms such as maturity-onset diabetes of the young4 and mitochondrial diabetes5 or late-onset autoimmune diabetes of the adult, which is actually a late-onset type 1 diabetes.6 Thus, most diabetes in the world is accounted for by "common" type 2 diabetes, which has a multifactorial pathogenesis caused by alterations in several gene products.
The medical and socioeconomic burden of the disease is caused by the associated complications,7-9 which impose enormous strains on health-care systems. The incremental costs of patients with type 2 diabetes arise not only when the diagnosis is established but at least 8 years earlier.10 The devastating complications of diabetes mellitus are mostly macrovascular and microvascular diseases as a consequence of accelerated atherogenesis. Cardiovascular morbidity in patients with type 2 diabetes is two to four times greater than that of non-diabetic people.1
Diagnosis
Diabetes mellitus is diagnosed on the basis of WHO recommendations from 1999, incorporating both fasting and 2-h after glucose load (75 g) criteria into a practicable diagnostic classification that should now be used (table 1).11 Conditions that predispose to overt diabetes, including impaired fasting glucose and impaired glucose tolerance are not merely of academic interest, since, unless treated, about 7% of people with these problems will progress to overt diabetes every year.12,13 Furthermore, impaired glucose tolerance itself carries an increased risk of macrovascular disease.14

Heredity in type 2 diabetes mellitus
Although lifestyle and overeating seem to be the triggering pathogenic factors, genetic elements are also involved in the pathogenesis of type 2 diabetes. Positive family history confers a 2·4 fold increased risk for type 2 diabetes. 15-25% of first-degree relatives of patients with type 2 diabetes develop impaired glucose tolerance or diabetes.15 The lifetime risk (at age 80 years) for type 2 diabetes has been calculated to be 38% if one parent had type 2 diabetes.15 If both parents are affected, the prevalence of type 2 diabetes in the offspring is estimated to approach 60% by the age of 60 years.16
Since dizygotic twins share the environment (both intrauterine and extrauterine) but only 50% of their genes, concordance rates in monozygotic twins in excess of those in dizygotic twins have been used to distinguish genetic from non-genetic contributions. In individuals older than 60 years, concordance rates for diabetes were 35-58% in monozygotic twins, compared with 17-20% in dizygotic twins.17,18 Inclusion of impaired glucose tolerance markedly increased the concordance in monozygotic twins to 88%.19
Nevertheless, concordance rates in monozygotic twins might produce an underestimate of genetic effects, because the monochorionic intrauterine nutrition of monozygotic twins has been shown to result in growth retardation compared with dizygotic twins.32 And low birthweight itself is associated with increased risk of type 2 diabetes later in life. 33,34
Pathophysiology of hyperglycaemia
To understand the cellular and molecular mechanisms responsible for type 2 diabetes it is necessary to conceptualise the framework within which glycaemia is controlled. Insulin is the key hormone for regulation of blood glucose and, generally, normoglycaemia is maintained by the balanced interplay between insulin action and insulin secretion. Importantly, the normal pancreatic ßcell can adapt to changes in insulin action--ie, a decrease in insulin action is accompanied by upregulation of insulin secretion (and vice versa). Figure 2 illustrates the curvilinear relation between normal ß-cell function and insulin sensitivity.35 Deviation from this hyperbola, such as in the patients with impaired glucose tolerance and type 2 diabetes in figure 2, occurs when ß-cell function is inadequately low for a specific degree of insulin sensitivity. Thus, ß-cell dysfunction is a critical component in the pathogenesis of type 2 diabetes. This concept has been verified not only in cross-sectional studies but also longitudinally in Pima Indians progressing from normal to impaired glucose tolerance to type 2 diabetes.36
In people with normal glucose tolerance (NGT) a quasi-hyperbolic relation exists between ß-cell function and insulin sensitivity. With deviation from this hyperbola, deterioration of glucose tolerance (impaired glucose tolerance [IGT], and type 2 diabetes [T2DM]) occurs.
However, not only deviation from but also progression along the hyperbola affects glycaemia. When insulin action decreases (as with increasing obesity) the system usually compensates by increasing ß-cell function. However, at the same time, concentrations of blood glucose at fasting and 2 h after glucose load will increase mildly.37 This increase may well be small, but over time becomes damaging because of glucose toxicity, and in itself a cause for ß-cell dysfunction. Thus, even with (theoretically) unlimited ß-cell reserve, insulin resistance paves the way for hyperglycaemia and type 2 diabetes.
Insulin resistance
Insulin resistance is said to be present when the biological effects of insulin are less than expected for both glucose disposal in skeletal muscle and suppression of endogenous glucose production primarily in the liver.38 In the fasting state, however, muscle accounts for only a small proportion of glucose disposal (less than 20%) whereas endogenous glucose production is responsible for all the glucose entering the plasma. Endogenous glucose production is accelerated in patients with type 2 diabetes or impaired fasting glucose.39,40 Because this increase occurs in the presence of hyperinsulinaemia, at least in the early and intermediate disease stages, hepatic insulin resistance is the driving force of hyperglycaemia of type 2 diabetes (figure 3).
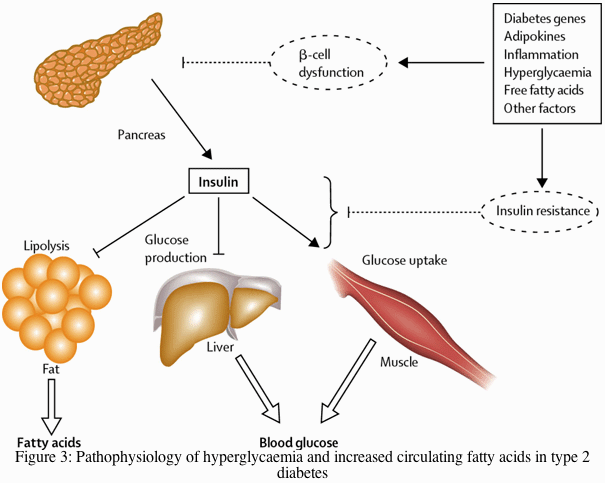
Insulin secretion from the pancreas normally reduces glucose output by the liver, enhances glucose uptake by skeletal muscle, and suppresses fatty acid release from fat tissue. The various factors shown that contribute to the pathogenesis of type 2 diabetes affect both insulin secretion and insulin action. Decreased insulin secretion will reduce insulin signalling in its target tissues. Insulin resistance pathways affect the action of insulin in each of the major target tissues, leading to increased circulating fatty acids and the hyperglycaemia of diabetes. In turn, the raised concentrations of glucose and fatty acids in the bloodstream will feed back to worsen both insulin secretion and insulin resistance.
Obesity
Insulin resistance is strongly associated with obesity and physical inactivity, and several mechanisms mediating this interaction have been identified. A number of circulating hormones, cytokines, and metabolic fuels, such as non-esterified (free) fatty acids (NEFA) originate in the adipocyte and modulate insulin action. An increased mass of stored triglyceride, especially in visceral or deep subcutaneous adipose depots, leads to large adipocytes that are themselves resistant to the ability of insulin to suppress lipolysis. This results in increased release and circulating levels of NEFA and glycerol, both of which aggravate insulin resistance in skeletal muscle and liver (figure 3).41
Excessive fat storage not only in adipocytes but "ectopically" in non-adipose cells also has an important role.42 For example, increased intramyocellular lipids are associated with skeletal muscle insulin resistance under some circumstances.43 The coupling between intrahepatic lipids and hepatic insulin resistance seems to be even tighter.44,45
Insulin receptor knock-out models
To understand the contribution of insulin resistance in a particular tissue to whole body glucose homoeostasis, conditional knockouts of the insulin receptor have been created using the Cre-lox system. Among the five conditional insulin receptor knockouts shown in table 3, only liver47 and ß-cell specific knockouts50 became glucose intolerant whereas, unexpectedly, knockout models specific for muscle46 and fat cells48 did not. These findings clearly support a central role of hepatic insulin resistance in the pathogenesis of type 2 diabetes, and suggest that an adequate insulin signal in the pancreatic ß cell is needed to maintain its function.

Cellular mechanisms
Insulin elicits its pleiotropic metabolic responses by binding to and activating a specific plasma membrane receptor with tyrosine kinase activity.51 Cellular substrates of the insulin receptor kinase, most prominently the insulin receptor substrate (IRS) proteins, are efficiently tyrosine phosphorylated on several sites, which serve as binding scaffolds for various adaptor proteins and lead to the downstream signalling cascade (figure 4).52 Insulin activates a series of lipid and protein kinase enzymes linked to the translocation of glucose transporters to the cell surface, synthesis of glycogen, protein, mRNAs, and nuclear DNA, which affect cell survival and proliferation.
Phosphorylation and dephosphorylation of IRS proteins
In states of insulin resistance, one or more of the following molecular mechanisms to block insulin signalling are likely to be involved. The positive effects on downstream responses exerted by tyrosine phosphorylation of the receptor and the IRS proteins are opposed by dephosphorylation of these tyrosine side-chains by cellular protein-tyrosine phosphatases and by protein phosphorylation on serine and threonine residues, which often occur together.53 Phosphotyrosine phosphatase 1B (PTP1B) is widely expressed and has an important role in the negative regulation of insulin signalling.54
Insulin signalling involves binding of insulin to its receptor followed by a cascade of intracellular events, depicted as activation pathways. Negative modulation of insulin action can be mediated via various pathways leading to insulin resistance: various inhibitory triggers affect their respective signal modulators (partly via transcription factors), which lead through deactivating pathways (tyrosine phosphatases, serine kinases, lipid phosphatases and degradation pathways) to inhibitory actions on insulin signalling (activation pathways). Adiponectin has an ameliorating function on glucose metabolism apart from insulin signalling. PKC=protein kinase C. PTEN=phosphatase and tensin homologue. PI=phospho-inositol.
Serine/threonine phosphorylation of IRS1 reduces its ability to act as a substrate for the tyrosine kinase activity of the insulin receptor and inhibits its coupling to its major downstream effector systems. Several IRS serine kinases have been identified, including various mitogen-activated protein kinases, c-Jun NH2-terminal kinase, atypical protein kinase C, and phosphatidylinositol 3-kinase, among others.52 Signal downregulation can also occur through internalisation and loss of the insulin receptor from the cell surface and degradation of IRS proteins.55 Members of the suppressor of cytokine signalling (SOCS) family of proteins participate in IRS protein degradation through a ubiquitin-proteosomal pathway (figure 4).56
Role of adipocyte products and inflammation
Increased concentrations of NEFA and inflammatory cytokines (eg, tumour necrosis factor alpha [TNFalpha] and interleukin 6) released by expanded visceral adipose tissue adversely affect the insulin signalling cascade.57,58 NEFA inhibit insulin-stimulated glucose metabolism in skeletal muscle and stimulate gluconeogenesis in liver.59,60 They activate cellular kinases, including atypical protein kinase C isoforms by increasing cellular diacylglycerol levels, which can activate the inflammatory kinases inhibitor kappaB kinase (IKK) and c-jun N-terminal kinase, increasing serine/threonine phosphorylation of IRS1 and reducing downstream IRS1 signalling, as previously described.61-63 TNFalpha enhances adipocyte lipolysis, which further increases NEFA, and also elicits its own direct negative effects on insulin signalling pathways.64 Neutralisation of TNFalpha substantially reverses insulin resistance in rodents; however, the magnitude of its involvement in human insulin resistance is not entirely clear.65 The proinflammatory interleukin 6 inhibits the insulin signal by augmenting the expression of SOCS proteins.66,67
Adiponectin
Whereas circulating NEFA and several adipokines are increased in visceral obesity, the concentrations of the adipose-specific protein adiponectin are decreased, reducing its insulin-sensitising effects in liver and muscle.57,68 Adiponectin signals via AMP kinase, a stress-activated signalling enzyme implicated in various metabolic responses, including suppression of hepatic gluconeogenesis, glucose uptake in exercising skeletal muscle, fatty acid oxidation, and inhibition of lipolysis, which might explain its beneficial metabolic effects.68-72 AMP kinase has also been implicated in the mechanism of action of metformin,73,74 and possibly of the thiazolidinediones,75 suggesting that it has a role in clinical anti-diabetic responses.
NFkappaB and IKK activity
A close connection between insulin resistance and classic inflammatory signalling pathways has also recently been identified. Nuclear factor kappaB (NFkappaB) is held in an inactive state in resting conditions by binding to an inhibitory partner, IkappaB.76 Phosphorylation of IkappaB by its kinase (IKK) leads to IkappaB degradation, releasing NFkappaB for translocation to the nucleus where it can affect the transcription of diverse genes involved in the inflammatory response. High doses of salicylates, which block IKK activity,77 can ameliorate hyperglycaemia and insulin resistance in diabetes and obesity.78,79 More importantly, genetic disruption of IKKß returned skeletal muscle insulin resistance caused by NEFA to normal, through improvement in IRS1 tyrosine phosphorylation and activation of its downstream signal cascade.80 Overall, this evidence suggests that IKK might be an important target for the development of new therapeutics in insulin resistance, especially in the setting of visceral adiposity.
In addition to their effects on insulin signalling, the circulating adipose tissue factors strongly affect vascular endothelial function, linking the increased vascular risk in the metabolic syndrome with mechanisms of cellular insulin resistance.68,81 Adipose secretory factors also recruit and activate inflammatory cells, which can further perpetuate a systemic inflammatory milieu that can strongly affect vascular function and atherogenesis.82
Mitochondrial metabolism
The accumulation of ectopic triglyceride in visceral depots (mainly in the liver) has suggested a defect in mitochondrial lipid oxidation in patients with type 2 diabetes, who have impaired oxidative capacity and small mitochondria in skeletal muscle.83 PPARgamma co-activator 1 (PGC1), a transcription factor for genes involved in mitochondrial fatty acid oxidation and ATP synthesis, was decreased in young, lean, insulin-resistant offspring of parents with type 2 diabetes, suggesting that an inherited defect in mitochondrial oxidative phosphorylation could lead to cellular lipid accumulation.84 Gene expression profiling studies have also shown that decreased expression of PGC1 and related gene products could affect mitochondrial function in people with insulin-resistance and type 2 diabetes.85,86
ß-cell dysfunction
Various abnormalities in insulin secretion are present in patients with type 2 diabetes. Basal insulin concentrations may be raised to roughly double the usual value, especially in obese hyperglycaemic patients, but this finding is presumably due to increased plasma glucose. Similarly, after a meal, concentrations of insulin in plasma can appear higher than normal, because of substantially raised plasma glucose. Indeed, hyperglycaemic glucose clamp studies under standardised conditions of identical glycaemia have shown that insulin secretion is markedly diminished compared with non-diabetic individuals with similar anthropometric characteristics.87 But even before the development of overt hyperglycaemia, which in itself adversely affects ß-cell function, secretory defects have been shown, for example in individuals with impaired glucose tolerance and impaired fasting glucose.88 Moreover, both normoglycaemic offspring of type 2 diabetic parents and non-diabetic twin-siblings of diabetic patients have reduced insulin secretion.87 Thus, in predisposed individuals, an insulin secretory defect is present, possibly on a genetic basis.89 Obesity, acute illness, or simply ageing might further expose or aggravate the underlying defect, ultimately leading to overt diabetes.87
In Pima Indians, a group known for their insulin resistance, prospective studies have shown that low insulin release determines the transition from normal to impaired glucose tolerance; conversely, Pima Indians who do not progress to the diabetic state were found to be able to increase their insulin secretion over time.36
Normal insulin secretion
Glucose is rapidly taken up by the pancreatic ß cell via the glucose transporter 2 (GLUT2), upon which it is phosphorylated via glucokinase, which is the rate-limiting step of ß-cell glucose metabolism (figure 5). Further degradation leads to formation of pyruvate, which is then taken up in the mitochondria in which further metabolism leads to ATP formation. ATP is necessary for the delivery of energy needed for the release of insulin, but it is also involved in the cell membrane depolarisation. The ADP/ATP ratio leads to activation of the sulphonylurea receptor 1 (SUR1) protein which will lead to closure of the adjacent potassium channel (potassium inward rectifier [KIR] 6·2 channel). The closure of the potassium channels will alter the membrane potential and open calcium channels, which triggers the release of preformed insulin-containing granules (figure 5).
Glucose toxicity
The notion that hyperglycaemia itself can decrease insulin secretion has led to the concept of glucose toxicity, which implies the development of irreversible damage to cellular components of insulin production over time.90,91 Indeed, deterioration of insulin secretion over time is the usual course in most patients, and many patients will end with more or less severe insulin deficiency after about 10 years of diabetes.92
In ß cells, oxidative glucose metabolism will always lead to production of reactive oxygen species, normally detoxified by catalase and superoxide dismutase. ß cells are equipped with a low amount of these proteins and also of the redox-regulating enzyme glutathione peroxidase.90 Hyperglycaemia has been proposed to lead to large amounts of reactive oxygen species in ß cells, with subsequent damage to cellular components (figure 6). Loss of pancreas duodenum homeobox 1 (PDX-1), a critical regulator of insulin promoter activity, has also been proposed as an important mechanism leading to ß-cell dysfunction.90 The finding in the diabetes-prone Zucker Diabetic Fatty rat that loss of pancreas duodenum homeobox-1 could be diminished by various manoeuvres preventing hyperglycaemia supports this. Additionally, reactive oxygen species are known to enhance NFkappaB activity, which potentially induces ß-cell apoptosis.
Lipotoxicity
More recently, the concept of lipotoxicity involving the ßcell has been put forward. Generally, in both non-diabetic and diabetic obese patients, NEFA concentrations are raised as a result of enhanced adipocyte lipolysis. Fatty acids lead to enhanced insulin secretion in acute studies, but after 24 h they actually inhibit insulin secretion. In the presence of glucose, fatty acid oxidation in ß cells is inhibited and accumulation of long-chain acyl coenzyme A occurs.93 This mechanism has been proposed to be an integral part of the normal insulin secretory process. However, long-chain acyl coenzyme A itself can also diminish the insulin secretory process by opening ß-cell potassium channels (figure 6). A second mechanism might be increased expression of uncoupling protein-2, which would lead to reduced ATP formation and, hence, decreased insulin secretion. A third mechanism might involve apoptosis of ß cells, possibly via fatty acid or triglyceride-induced ceramide synthesis or generation of nitric oxide.
Islet amyloid
Islet amyloid consists of deposits of islet amyloid polypeptide, also known as amylin, which is co-secreted with insulin at a more than tenfold lower rate. The physiological role of islet amyloid polypeptide is unclear, and diverse roles such as inhibition of insulin action, inhibition of insulin secretion, and inhibition of glucagon secretion have been proposed. It has been suggested that small aggregates are cytotoxic,94 possibly related to radical production. NEFA may add to the cytotoxicity of the aggregates.95 Amyloid deposits are found in most but not all individuals with type 2 diabetes.96
It is possible that early in the disease, increased demands of insulin secretion lead to islet amyloid polypeptide aggregates, especially in the presence of raised concentrations of NEFA. The finding that first-degree relatives of patients with type 2 diabetes have decreased islet amyloid polypeptide (and insulin) responses to intravenous glucose, however, challenges this speculation.96 Also, amyloid is not observed in middle-aged insulin-resistant individuals. Thus, the role of amyloid deposits (a post-mortem finding) in pancreatic islets in the pathophysiology of type 2 diabetes remains unclear.
Genetic factors
Although there is little doubt as to the importance of genetic factors in type 2 diabetes (table 2), it should be borne in mind that this disease is very heterogeneous. Genetic studies have therefore given very diverse results. In general, two methods are used for studying genetic factors involved in a specific disease: the so-called candidate gene approach and the genome-wide scan approach.
The candidate gene approach examines specific genes with a plausible role in the disease process. For this purpose the statistical association of a given allele and a phenotype (eg, type 2 diabetes, or insulin resistance) is tested in unrelated individuals. The genome-wide scan or linkage approach is not based on assumptions but locates genes through their genomic position and is based on the rationale that family members sharing a specific phenotype will also share chromosomal regions surrounding the gene involved.
Candidate genes
The candidate gene approach in attempts to identify a causative factor among the obvious biological candidates for insulin resistance has been largely disappointing (table 4). Variants in many candidate genes were extensively studied over the past two decades, such as the Gly972Arg polymorphism in IRS1, the Gly1057Asp polymorphism in IRS2, the Trp64Arg polymorphism in the ß3 adrenergic receptor, the -308 G/A promoter variant in TNFalpha, or variants in the adiponectin gene. In most instances the initial association was not replicated in subsequent analyses and, currently, the most robust single candidate variant is the highly prevalent Pro12Ala polymorphism in peroxisome proliferator-activated receptor gamma (PPARgamma).97,98
PPARgamma
PPARgamma is a transcription factor that is activated by certain fatty acids, prostanoids, and thiazolidinediones.99,100 Whereas the isoform PPARgamma1 is expressed in most tissues, PPARgamma2 is specific for adipose tissue, where it has a key role in regulation of adipogenic differentiation.101 The high risk proline allele of the Pro12Ala PPARgammapolymorphism has a prevalence of 75% in white people. In 333 Scandinavian parent-offspring trios with abnormal glucose tolerance, among 16 candidate gene variants only the P12A polymorphism in the PPARgamma gene (PPARG) was significant.102 Results of two meta-analyses and a large prospective analysis have shown a risk reduction between 21% and 27% for the alanine allele.97,98,103 The alanine genotype presumably results in greater insulin sensitivity.104-106 The proline variant has lower transcriptional activity and heterozygous PPARgammaknockout mice are more insulin resistant. Since PPARgamma2 is exclusively expressed in adipose tissue, a primary mechanism in this tissue with a secondary effect on hepatic insulin sensitivity and insulin clearance can be invoked.107 Interestingly, in Pima Indians, insulin suppression of glucose production was 40% more efficient in carriers of the Ala allele whereas insulin-stimulated glucose uptake did not differ.106
IRS1 and PGC1alpha
The Gly972Arg polymorphism in IRS1, a variant intuitively associated with insulin resistance, may have a weak association with type 2 diabetes,97 although possibly through ß-cell dysfunction rather than insulin resistance.108,109 The Gly483Ser polymorphism in PGC1alpha, a transcriptional co-factor, might also be associated with type 2 diabetes via as yet unknown mechanisms.110
Sulphonylurea receptor-1/potassium inward rectifier 6·2
Among the many candidate genes for insulin secretory dysfunction, those encoding SUR1 and KIR6·2 have been most extensively studied. The two genes--ABCC8 and KCNJ11, respectively--are adjacent to one another on chromosome 11. There is insufficient evidence for association of two widely studied SUR1 polymorphisms (exon 16-3t/c, exon 18 T759T) with type 2 diabetes.111 Meta-analyses on the E23K variant in the KIR6·2 gene are more robust, suggesting that the risk of type 2 diabetes increased by about 15% for the K allele,111 probably through decreased insulin secretion. A recent haplotype analysis using an independent dataset confirmed the association with the KIR6·2 variant and further substantiated the notion that genetic variation in the SUR1/KIR6·2 region is associated with type 2 diabetes.112 However, because the E23K polymorphism is in strong linkage disequilibrium with a nearby coding variant in the SUR1 gene (A1369S), it is difficult to distinguish the roles of the two polymorphisms.
Insulin-like growth factor
Genes involved in embryonic ß-cell development, such as components of the insulin-like growth factor pathways, have also been studied. Although intact insulin/insulin-like growth factor signalling is clearly essential for ß-cell growth and development,113 the potential role of genetic variants remains an area of research.
Genome-wide scans
Several findings of positive associations of genomic regions with type 2 diabetes have been replicated in one or more studies (1q21-24, 1q31-q42, 9q21, 10q23, 11p15, 11q13-14, 12q12, 19q13, and 20q11-q13 ).114 Generally, such findings are followed by positional cloning of the causative gene, which to date has not been successful for most regions.
Calpain-10
The first "common diabetes gene" cloned in this way was CAPN10 in the NIDDM1 region of chromosome 2.115,116 It encodes for calpain-10, a cysteine protease which is ubiquitously expressed.117,118 Originally, the G allele of a non-coding single nucleotide polymorphism (UCSNP-43) was reported to be associated with type 2 diabetes, while specific haplotype combinations of several single nucleotide polymorphisms (UCSNP-19, 43, 63) seemed to increase the risk of diabetes in selected populations.116 In later studies, this finding could not be replicated119,120 but another single nucleotide polymorphism (UCSNP-44) was found to be associated with type 2 diabetes.121-124 Genetic variants in calpain-10 might affect insulin sensitivity,125 or insulin secretion,126 or the relation between the two (table 4).127
HNF4A
Abnormalities in the HNF4A gene cause maturity-onset diabetes of the young type 1. Genetic variation near or in the P2-promoter of the MODY-1 gene HNF4A gene (chromosome 20q) has been proposed to relate to common type 2 diabetes,128 but this finding requires independent confirmation.
Insulin gene variable number tandem repeat
A peculiar possibility is the relation of diabetes to imprinted genes--ie, genes for which expression varies depending on the sex of the transmitting parent. The class III allele of the variable number tandem repeat near the insulin gene (chromosome 11p15) might relate to type 2 diabetes.129 The class III allele is associated with decreased amounts of insulin mRNA. Only paternally transmitted class III alleles were found to be associated with diabetes in one study.130
Management of hyperglycaemia
In making therapeutic choices (figure 7) in the management of type 2 diabetes, the major goal of protecting patients from the long-term complications of the disease must be considered. Because insulin resistance plays a fundamental role in the pathogenesis of type 2 diabetes and especially its adverse cardiovascular outcomes, interventions should initially be aimed towards improvement in tissue insulin sensitivity. This often involves lifestyle intervention, with modest exercise and weight loss, which clearly reduces the risk of progression of impaired glucose tolerance to overt diabetes12,13 and can improve many of the cardiovascular risk parameters of the metabolic syndrome.
Thiazolidinediones
Drugs that enhance insulin sensitivity are primarily those of the thiazolidinedione class, which not only reduce glycaemia, but also enhance vascular function and ameliorate the dyslipidaemia and inflammatory milieu of type 2 diabetes.131 Thiazolidinediones primarily activate PPARgamma receptors in adipose tissue and alter adipose metabolism and distribution. The redistribution of tissue triglyceride from visceral stores reduces levels of circulating NEFA apparently by sequestration in a less lipolytic subcutaneous compartment.132 Thiazolidinediones also reduce circulating concentrations of pro-inflammatory cytokines that promote insulin resistance (eg, TNFalpha and interleukin 6) and at the same time increase concentrations of adiponectin, which has insulin-sensitising and anti-inflammatory properties. The multiple effects of thiazolidinediones on adipose tissue metabolism and cross-talk of these signals with liver and skeletal muscle, as well as pancreatic ß cells and the vascular endothelium, might account for the enhancement of insulin action and improvement in insulin secretion with these agents, as well as several beneficial effects on vascular function.133 The action of the thiazolidinediones to redistribute visceral triglyceride can reduce hepatic lipid content in non-alcoholic steatohepatosis, which is closely related to obesity and insulin resistance.131 Ongoing studies will determine if these agents can reduce the risk of inflammation leading to cirrhosis in this condition. The renal and vascular benefits of thiazolidinediones have been demonstrated in controlled studies, for example, showing significant improvement in albumin excretion above that observed with a similar degree of glycaemic lowering with sulfonylureas.134
Unlike metformin, the thiazolidinediones can be used in patients with reduced renal function, and they are better tolerated without significant gastrointestinal side-effects. A major adverse effect associated with clinical use of the thiazolidinediones is weight gain, which seems to be coupled to the effects of the drugs on adipose cell differentiation and triglyceride storage. Fluid retention is also linked to the PPARgamma-agonist activity of the thiazolidinediones, leading to peripheral oedema and a mild haemodilution in some patients. Fortunately, congestive heart failure is quite rare with use of thiazolidinediones, but remains a serious concern that requires caution in selection of patients to receive these agents.135 The ability of thiazolidinediones to ameliorate risk of atherosclerotic events is being assessed in several large outcomes studies.
Metformin
Metformin is a highly effective antihyperglycaemic drug that works independently of the pancreas, sparing insulin. It decreases hepatic glucose output and has been shown to have a beneficial effect on cardiovascular outcomes.136-138 Metformin has less robust effects on insulin resistance, inflammatory markers, and vascular function compared with the thiazolidinediones, but its benefit in abrogating some of the weight gain commonly observed with insulin-sensitisers and insulin secretion enhancers adds important value to this drug.
alpha-glucosidase inhibitors
The alpha-glucosidase inhibitor acarbose has been shown to reduce glycaemic excursions and protect against the development of diabetes and cardiovascular disease.139
Sulfonylurea derivatives
As inadequate ß-cell insulin secretion is fundamental to the development of hyperglycaemia in diabetes, insulin secretion enhancers also play an important role in control of blood glucose. Sulfonylurea derivatives act by closing pancreatic cell potassium channels, which leads to enhanced insulin secretion. The results of the UK Prospective Diabetes Study8 showed a clear risk reduction for the occurrence of microvascular complications by the use of sulfonylurea derivatives, while the risk reduction of macrovascular disease was around 16%. Antihypertensive therapy diminished the risk of macrovascular complications by around 20%. Combined management with both sulfonylurea derivatives and antihypertensives improves the risk reduction even more.
The mode of action of sulfonylurea derivatives implies that they also act at low concentrations of plasma glucose, which explains the potential of (occasionally severe) hypoglycaemia. Whereas tolbutamide, gliclazide, and glipizide have a relatively short durations of action, glimepiride and glibenclamide (glyburide) are long acting (24 h), adding to the risk of hypoglycaemia.140 The risk of hypoglycaemia in the other sulfonylurea derivatives is lower, with a possible advantage of gliclazide in patients with decreased kidney function.141 Sulfonylurea derivatives will lead to moderate decreases in concentrations of plasma glucose in most patients with type 2 diabetes, and concentrations of glycosylated haemoglobin will generally decrease by about 1-2%.
The recently introduced class of meglitinides consists of nateglinide, which binds to the same site of sulphonylurea receptor 1 as do the sulfonylurea derivatives, and repaglinide, which binds to a nearby site of the receptor, both leading to insulin release. These agents cannot further stimulate insulin release in patients on maximal doses of sulfonylurea derivatives. Both agents have a shorter action than sulfonylurea derivatives, are therefore associated with lower risk of hypoglycaemia, and can also be used in patients with decreased renal function.140
Exogenous insulin
Replacing circulating concentrations of insulin is essential to support the clinical effects of metformin and the thiazolidinediones, which are ineffective without adequate insulin availability, and may also have important beneficial effects in reducing inflammatory processes, especially in the vasculature.142 Thus, it is essential to initiate insulin injections when required to achieve glycaemic targets in type 2 diabetes, possibly in combination with oral insulin sensitisers. However, combined use of insulin and thiazolidinediones seems to infer an increased risk of oedema and cardiac failure. Therefore, this combination is not allowed in most European countries.
Glucagon-like peptide 1
A novel insulin secretagogue concept has been built around glucagon-like peptide 1. This incretin hormone has potent glucose-dependent insulinotropic properties, trophic effects on ß cells, and inhibitory effects on intestinal motility, all of which reduce plasma glucose. However, because circulating glucagon-like peptide 1 is immediately inactivated by dipeptidyl peptidase IV, it is therapeutically impractical. Dipeptidyl peptidase IV-resistant analogues and selective dipeptidyl peptidase IV inhibitors have been developed and are in the final stages of approval (figure 7).143
Experimental approaches
As specific drug targets are identified through improved understanding of the molecular pathogenesis of diabetes, novel therapeutics will become available in the future. For example, PTP1B is a negative regulator of insulin signalling, and inhibition of its activity with specific pharmaceutical agents, or reduction of its protein concentrations with novel antisense oligonucoleotides, has been shown to enhance insulin action in pre-clinical models.144
Controlling excessive secretion of TNFalpha or interleukin 6 or blocking their action mediated by serine/threonine kinases would be expected to enhance insulin sensitivity in patients with visceral adiposity. Conversely, increasing adiponectin secretion or administration of an adiponectin receptor agonist would probably enhance glucose metabolism in skeletal muscle and liver and also confer beneficial effects in the endothelium. Recent evidence for amelioration of insulin resistance by salicylates by favourable interference with the inflammatory kinase cascade in insulin signalling might lead to entirely novel therapeutic approaches.
Management of other cardiovascular risk factors
A complete discussion of this topic is outside the scope of this paper and the metabolic syndrome is discussed in a separate Seminar.145 Hypertension, coronary artery disease, and cerebrovascular disease, occur more often in type 2 diabetes than in matched controls.146 The UK Prospective Diabetes Study8 has convincingly shown the benefit of vigorous antihypertensive therapy in patients with type 2 diabetes. The benefit of antihypertensive therapy is larger in diabetic than in non-diabetic hypertensive patients. To lower blood pressure to the levels recently proposed by the American Diabetes Association (less than 130/80 mm Hg), many patients will need two or three different antihypertensive drugs. ßblockers, diuretics, angiotensin-converting enzyme (ACE) inhibitors, calcium channel antagonists, and angiotensin receptor blockers can all effectively decrease blood pressure in type 2 diabetes, while alpha blockers are probably less effective.146
ACE inhibition
The results of the HOPE study,147 in which use of ramipril was associated with a markedly lower risk of myocardial infarction, stroke, and death, favour use of the ACE inhibitor in diabetic patients with one additional risk factor, even if they do not have hypertension. Losartan may have an advantage over the ß blocker atenolol in decreasing cardiovascular mortality.146 Whether angiotensin receptor blockers will show greater benefit than ACE inhibitors, or whether these two classes of drugs should be used together, as suggested by lower blood pressure during combination therapy148 is yet to be ascertained.
Statins
The benefit of lipid-lowering drugs has now been firmly established, since the Scandinavian Simvastatin Survival Study showed a reduction in total mortality of 43%. Similarly, the Heart Protection Study showed a 25% lower risk in end points for simvastatin use.149 The Adult Treatment Panel III of the National Cholesterol Education Program in the USA sets the LDL-cholesterol goal at less than 2·6 mmol/L. It also aims for triglyceride concentrations of less than 1·7 mmol/L, and for HDL cholesterol concentrations of greater than 1·0 mmol/L.150
Fibric acid derivatives might benefit diabetic patients because they raise concentrations of HDL cholesterol and reduce triglyceride levels. They can decrease endothelial cell activation possibly because of their capacity for binding PPARalpha. They have been shown to reduce the risk for myocardial infarction by 24%.146 Nicotinic acid might also raise HDL cholesterol, but could lead to higher glycaemia.151
Aspirin
The management of other risk factors remains under debate. Antiplatelet treatment (generally aspirin) has been shown to decrease the risk of atherosclerotic manifestations by 19%.146 The American Diabetes Association recommends to prescribe low-dose aspirin as a secondary prevention strategy after manifestations of atherosclerotic disease, and to use aspirin as primary prevention in patients with a high risk for atherosclerotic disease, especially patients older than 40 years, or with an additional risk factor such as family history of cardiovascular disease, hypertension, smoking, dyslipidaemia or albuminuria.152 There is still uncertainty about the dose of aspirin that is needed.
Whether therapy should be given for other risk factors such as hyperhomocysteinaemia (treated with folic acid), and whether antioxidants are of use, is still unclear because of the absence of studies with hard endpoints in patients with type 2 diabetes.
Diabetes prevention
The goal of ultimately reducing the population burden of diabetes by early treatment and prevention is clearly of pivotal importance. A number of studies have shown that diabetes can be delayed or prevented in individuals at high risk undergoing an intensive diet and exercise programme, and intervention with medications including metformin, acarbose or thiazolidinediones has also shown to be effective (table 5). The interesting observation that improvement in one or more major pathogenic factors offsets the progression of impaired glucose tolerance to diabetes underscores the contribution of each of these factors to the development of the disease, including insulin sensitivity, ß-cell function, and actual glucose excursions. Thus, careful attention needs to be applied to determine appropriate public interventions for the varied populations of the world. Lifestyle modification has been difficult to maintain over a long term, and has costs associated with regular visits to various health-care professionals and lifestyle coaches. Medications may have unwanted side-effects. Further clinical trials comparing these and newer medications that may affect diabetes pathogenesis (such as glucagon-like peptide 1 analogues) are needed to balance safety and efficacy with the costs of these different agents in various regions.
Future aspects
A more complete understanding of the molecular mechanisms of diabetes will enable the identification of individuals at highest risk, which could lead to novel pharmacological concepts, risk stratification, and development of more targeted preventive measures. A long-term goal is to develop drugs that restore normoglycaemia by targeting specific pathogenic defects. One example would be to advance the thiazolidinedione concept to design compounds that could restore defects in individuals with a defective PPARgamma regulatory system. Similarly, the understanding of the role of the SUR1/KIR6·2 complex inß-cell dysfunction might foster the development of a new generation of sulphonylurea-like agents. Antisense inhibition of PTP1B, a tyrosine phosphatase, currently undergoing phase II trials, could become the treatment of choice for patients with a genetic variant in PTP1B.156 Generally, with an optimised risk-benefit ratio, patients who respond to treatment may also benefit from specific drugs in a preventive approach. Until that day, diet and exercise remain the pillars of prevention and treatment of type 2 diabetes. The implementation and sensible use of the available pharmacological agents, including insulin, and the management of other cardiovascular risk factors, remain the practical challenge to the clinician.