| |
Can We Cure HIV: "Depletion of latent HIV-1 infection in vivo: a proof-of-concept study"
|
| |
| |
.....Our findings show that 16–18 weeks' treatment with a standard clinical dose of valproic acid, in the setting of intensified HAART, produces a substantial decline in the frequency of replication-competent HIV in circulating resting CD4+ T cells.....
The Lancet August 13 2005; 366:549-555
Ginger Lehrman a, Ian B Hogue a, Sarah Palmer b, Cheryl Jennings c, Celsa A Spina d, Ann Wiegand b, Alan L Landay c, Robert W Coombs e, Douglas D Richman d, John W Mellors f, John M Coffin b, Ronald J Bosch g and David M Margolis
News of the Week
HIV/AIDS:
Report of Novel Treatment Aimed at Latent HIV Raises the 'C Word'
Jon Cohen
A report in the 13 August issue of The Lancet is roiling the world of AIDS research. It describes an unusual treatment of four HIV-infected people, and the authors suggest that the strategy may point the way to a "cure of HIV in the future." The paper, co-written by a prominent collection of AIDS researchers, ventures into terrain that few have explored: eradicating the virus from all infected cells throughout the body. "The drumbeat we've been hearing for the last 5 years is this can't be done," says the study's leader, virologist David Margolis of the University of North Carolina, Chapel Hill. "The level of skepticism is very high. And rightly so. But the data we've gotten make me more hopeful."
In the AIDS field, few dare to use the "c word," and its use here has stirred criticism. Even "the words 'on the way to a cure' are just so inappropriate," says Robert Gallo, head of the Institute of Human Virology in Baltimore, Maryland, where Margolis once worked. "I think that was really a serious mistake." Researchers are wary in part because they have been burned before: Early enthusiasm in 1996 about the power of anti-HIV drug cocktails--called highly active antiretroviral therapy (HAART)--led David Ho of the Aaron Diamond AIDS Research Center (ADARC) in New York City to famously propose that eradication might take only 2 to 3 years of treatment. The concept lost currency when it became clear that HIV hides out in a latent state in cellular reservoirs from which it is very hard to dislodge.
In the new study, Margolis and co-workers recruited four patients who for 2 years had fewer than 50 copies of HIV per milliliter of blood, the detection limit of the standard assay. The researchers first drew huge amounts of blood from each patient through a process called leukopheresis and, using a highly sensitive test that purports to detect a single copy of HIV per milliliter, sifted through hundreds of millions of resting CD4+ white blood cells--the main harbor for latent virus--to assess the size of each patient's viral reservoir. They then gave the patients a drug they believed would force their resting cells to spit out new copies of HIV, which theoretically exposes the cells to immune attack or self-destruction. To help mop up bursts of the virus, the patients added a new drug, T-20, to their standard cocktails. After 4 months, the amount of infectious HIV in each patient's pool of latent cells declined an average of 75%, the investigators reported.
Researchers roundly praise Margolis for doing this difficult study, but reactions to the data and the postulated mechanism of action have been decidedly mixed. "It's an alternative approach that's worth pursuing," says Anthony Fauci, head of the National Institute of Allergy and Infectious Diseases in Bethesda, Maryland. "But you have to be very careful about the hope you have for eradication with this. We went through the same thing a few years ago." Fauci, working with Tae-Wook Chun in his lab, found that they could reduce the size of the pool of latently infected cells in two patients, using interleukin-2 (IL-2) to "activate" the resting cells and thus flush the latent virus out of hiding. But as Fauci, Chun, and colleagues reported in the 28 October 1999 issue of Nature, when the patients stopped treatment, HIV quickly surfaced and refilled latent pools within weeks. Other groups have reported similar results.
The new study takes a more precise approach. Drugs such as IL-2 that nonspecifically activate white blood cells also create more CD4+ cells for the virus to infect. "You're always going to be chasing your tail" with such strategies, says Margolis. Along with Carine Van Lint of the University of Brussels and Eric Verdin of the Gladstone Institute of Virology and Immunology in San Francisco, Margolis has evidence that an enzyme called histone deacetylase 1 (HDAC1) plays a crucial role in keeping CD4+ cells in a latent state. So Margolis gave his patients valproic acid, an HDAC1 inhibitor that's licensed to treat epileptic seizures. "It's more of a scalpel than the blunter instruments that we and others have used," agrees ADARC's Ho. "There are caveats about this study, but I'm certainly pleased to see their results."
Virologist Robert Siliciano of Johns Hopkins University in Baltimore stresses that even the 75% reduction of the latent pool has no clinical relevance. "Partial reductions of these cells sound good, but it's got to be complete to be useful," says Siliciano, whose lab specializes in HIV latency. He notes, too, that he and others disagree with Margolis about the mechanism of latency, questioning the value of HDAC1 inhibitors. "It's likely that there are several mechanisms," counters Margolis, who says this is just a small "proof of concept" study. Other researchers also caution that the assays used by Margolis and his collaborators are highly experimental, and it's unclear whether valproic acid or the intensification of HAART with T-20 were critical factors.
Roger Pomerantz, who also did eradication experiments in patients before he left academia to become president of the drug company Tibotec, says he's intrigued by the HDAC1 inhibitors, and he hopes the work spurs other clinical studies. Ho, who has taken heat for his earlier optimism, agrees. "It's OK to think about curing HIV," he says. "If we give up, there will never be a cure."
COMMENTARY in The Lancet
Valproic acid: a potential role in treating latent HIV infection
Highly active antiretroviral therapies (HAART) are dramatically improving HIV treatment outcomes, by shutting down virus production. However, this success has been incomplete because of chronic low levels of virus replication and because HIV genomes can persist silently in some infected cells in patients receiving HAART.1 As well as suppressing or controlling infection, eradicating HIV requires removing these residual, latently infected cells or flushing out the viral genomes integrated into them. Recent studies suggest that purging these cells from the body or flushing virus from infected cells is feasible.2
In today's Lancet, Ginger Lehrman and colleagues present their results of a proof-of-concept study, designed to assess the efficacy of a standard clinical dose of valproic acid to deplete HIV from resting CD4+ cells.3 After infection with HIV, CD4+ cells are activated and become highly susceptible to the virus because of the expression of CCR5, a co-receptor for HIV entry. As a consequence of activation, most of these cells, infected or not, will induce their own death through apoptosis. However, some infected CD4+ cells do survive and return from an activated state to a resting one.4 Then, HIV genomes integrated in these cells remain indefinitely present as a reservoir capable of HIV replication.5 This reservoir of dormant cells, predominately in the digestive mucosa, is not eradicated even after either HAART intensification or treatment with T-cell activators, such as interleukin 2.6 Now, there are promising results that drugs might selectively induce the expression of latent proviral genomes, and potentially deplete these reservoirs.
One of these drugs is valproic acid, a well-known anticonvulsant. It has been found to act as a histone deacetylation inhibitor regulating gene expression.7 Through its effect on chromatin structure, valproic acid is able to induce HIV expression in the resting T cells of aviraemic HAART-treated patients.8 Furthermore, this in-vitro effect has been observed in at least one other viral infection. B-cell lymphoma cells associated with infection with human herpesvirus 8 (primary effusion lymphoma) were selectively killed by apoptosis, without an increase in production of human herpesvirus 8, when incubated with valproic acid and the antiherpetic drug ganciclovir.9
In Lehrman and colleagues' study, four adults whose HIV infection was well-controlled with HAART were also treated with the fusion-entry inhibitor enfuvirtide, for 4–6 weeks before starting valproic acid. This was done to prevent the spread of virus in the presence of valproic acid. Responses were measured by the change in the number of infected cells per billion resting CD4+ cells and by an ultrasensitive assay capable of detecting as little as one copy of HIV RNA per mL plasma. The number of HIV-infected cells, measured twice before the start of valproic acid, was steady in all four patients. In three patients, the number of infected cells declined substantially 16–18 weeks after the start of valproic acid. The number of these infected cells subsequently rebounded after drug discontinuation. Furthermore, plasma HIV RNA levels did not change significantly in any patients during the study. The only side-effect attributable to valproic acid was zidovudine-related anaemia, probably explained by the increased bioavailability of zidovudine in the presence of valproic acid.
These results, although preliminary, merit further urgent study. In particular, they argue for a randomised trial to assess changes in the blood and tissue of the number of latently infected cells. As with most research, these results pose new questions. Can the virological effect be explained by a pharmacokinetic interaction between valproic acid and HAART?10 Are there dose-related effects? Are there other drugs to be studied? Might cytokines such as interleukin 7, or therapeutic vaccines, enhance this effect?11 Ultimately, more information is needed to characterise HIV-infected CD4+ memory subsets better in different tissue compartments, and to tease out the relative contribution of valproic acid on apoptosis of latently infected cells and on the removal of integrated HIV genomes from these cells.12 This is the first glimpse of a new therapeutic approach that might represent a possible step towards making HIV-infection no longer a chronic disease.
I declare that I have no conflict of interest.
STUDY TEXT
Summary
Background
Persistent infection in resting CD4+ T cells prevents eradication of HIV-1. Since the chromatin remodeling enzyme histone deacetylase 1 (HDAC1) maintains latency of integrated HIV, we tested the ability of the HDAC inhibitor valproic acid to deplete persistent, latent infection in resting CD4+ T cells.
Procedures
We did a proof-of-concept study in four volunteers infected with HIV and on highly-active antiretroviral therapy (HAART). After intensifying the effect of HAART with subcutaneous enfuvirtide 90 μg twice daily for 4–6 weeks to prevent the spread of HIV, we added oral valproic acid 500–750 mg twice daily to their treatment regimen for 3 months. We quantified latent infection of resting CD4+ T cells before and after augmented treatment by limiting-dilution culture of resting CD4+ T cells after ex-vivo activation.
Findings
The frequency of resting cell infection was stable before addition of enfuvirtide and valproic acid, but declined thereafter. This decline was significant in three of four patients (mean reduction 75%, range 68% to >84%). Patients had slight reactions to enfuvirtide at the injection site, but otherwise tolerated treatment well.
Interpretation
Combination therapy with an HDAC inhibitor and intensified HAART safely accelerates clearance of HIV from resting CD4+ T cells in vivo, suggesting a new and practical approach to eliminate HIV infection in this persistent reservoir. This finding, though not definitive, suggests that new approaches will allow the cure of HIV in the future.
Introduction
Antiretroviral therapy suppresses plasma HIV RNA concentrations below the limits of detection and restores immune function. However, latent replication-competent provirus in resting CD4+ T lymphocytes1–3 and persistent viral replication4,5 prevent eradication of HIV infection. Nevertheless, since lifelong suppression of HIV infection with antiretroviral therapy is difficult, eradication should be the therapeutic aim. Approaches to deplete latent infection within resting CD4+ T cells are, therefore, needed.
Results of a long-term study,6 which followed-up patients for up to 7 years, confirm the stability of latent infection. Although a modest increase in the decay of the number of recoverable cells that contain replication-competent HIV has been reported after intensified antiretroviral therapy,7 the rate of decay of plasma HIV RNA8 or integrated HIV DNA decreases with time.9,10 As such, a proportion of the pool of latently infected cells is probably highly stable and unlikely to be affected by potent inhibition of viral replication.
Intensive antiretroviral therapy in combination with T-cell activation does not eradicate HIV infection.11–14 Activation can induce viral replication and increase the number of susceptible uninfected target cells beyond the threshold that can be contained by antiretroviral therapy.15 The discovery of reagents that selectively induce the expression of quiescent proviral genomes, but that have limited activating effects, might allow outgrowth of latent HIV and avoid the pitfalls of global T-cell activation.16–19
Histone deacetylation is important for quiescence of HIV gene expression in infected resting CD4+ T lymphocytes. Histone deacetylase 1 (HDAC1) mediates chromatin remodeling, repression of viral gene expression, and virion production.20–23 Blockade of HDAC activity ex vivo results in HIV outgrowth from the resting T cells of aviraemic patients.23,24 Valproic acid, an anticonvulsant that inhibits HDAC, induces HIV expression ex vivo from the resting CD4+ T cells of aviraemic patients treated with highly-active antiretroviral therapy (HAART) as efficiently as activation with mitogen, but without upregulation of cell-surface markers of activation, or increased susceptibility to de-novo HIV infection.25
Our aim, therefore, was to test the ability of valproic acid to deplete HIV infection of resting CD4+ T cells in vivo.
Methods
Patients
Between July, 2002, and February, 2005, we did a proof-of-concept study to which we enrolled HIV-infected volunteers with long-term suppression of viraemia (<50 copies/mL for >2 years) who were taking HAART. Inclusion criteria were age 18 years or older, presence of documented HIV infection, ability to comply with protocol requirements and self-administer enfuvirtide for up to 18 weeks. Exclusion criteria were poor adherence to antiretroviral therapy, presence of contraindications to valproate therapy, receipt of interferon, other immunomodulators, or other experimental treatments.
All patients provided written informed consent, and the study was approved by the Dallas Veterans' Administration Medical Center institutional review board.
Procedures
We intensified the effect of HAART by concomitant subcutaneous administration of the fusion inhibitor enfuvirtide 90 μg twice daily after two rounds of leucopheresis. Once patients had adhered to HAART and enfuvirtide for 4–6 weeks, oral valproic acid 500–750 mg twice daily was initiated and continued for 3 months. We measured total and free serum concentrations of valproic acid, and did routine clinical haematology and blood chemistry monitoring once a week for a month, then monthly, and once again 1 month after protocol therapy was discontinued. We adjusted the dose of valproic acid to maintain plasma concentrations of 50–100 mg/L. We obtained plasma, peripheral blood mononuclear cells (PBMCs), and semen (when available) for analysis at scheduled intervals. Leucopheresis was done twice before the initiation of enfuvirtide and after 16–18 weeks' treatment with HAART, valproic acid, and enfuvirtide.
We obtained leucocytes by continuous-flow leucopheresis. The leucocytes were washed and separated from contaminating red cells by Ficoll gradient centrifugation. We isolated resting CD4+ T cells as described previously.2,24 FACS analysis confirmed greater than 99·9% purity of CD4+ cells with less than 0·5% of cells exhibiting activation markers. We obtained 200–1200 million resting CD4+ cells. After negative selection, we cultured the cells for 2 days in the presence of an integrase inhibitor (L-708906, 10 mmol/L; provided by D Hazuda, Merck Research Laboratory, West Point, PA, USA) and a selected reverse transcriptase inhibitor.26 We used efavirenz (15 nmol/L; provided by Bristol Meyers-Squibb, Wallingford, CT, USA) if the patient has not been treated with non-nucleoside reverse transcriptase inhibitors; alternatively we used abacavir (4 mmol/L, provided by Glaxo SmithKline, Research Triangle Park, NC, USA).
For the recovery and quantification of replication-competent HIV, we cultured 140–263 million resting cells in replicate dilution series of 5, 2·5, 1·25, and 0·625 million cells per well for every experimental condition.24 The serial dilution range was designed for optimum efficiency as per the statistical analysis of Macken.27,28
We induced the cells of patients to express HIV by exposure to 2 mg/L PHA-L (Remel, Lanexa, KS, USA), 6×106 allogeneic irradiated PBMCs, and 100 U/mL interleukin 2. As a control for low-level contamination of cultures with activated cells, we exposed parallel cultures to 100 U/mL interleukin 2 for 72 h. Cultures were subsequently fed and maintained as previously described.24 We scored cultures as positive if p24 was detectable 21 days after cell harvest, and rising p24 concentrations were confirmed on day 25. Virus was detected in culture supernatant by p24 antigen capture ELISA (Coulter, Miami, FL, USA).
We measured plasma HIV RNA concentrations by Roche Amplicor (Roche Molecular Systems, Branchburg, NJ, USA) with an assay detection limit of 50 copies per mL, and by a quantitative real-time, RT-PCR assay capable of detecting and quantifying HIV RNA concentrations of 1 copy per mL.29 We confirmed the ability of the primers and probe used to amplify and detect the viral RNA in every patient by testing of a sample of banked plasma taken before initiation of fully suppressive HAART. Since such a sample was not available for patient 3, we tested primers and probes on viral RNA recovered from the outgrowth assay of that patient's resting T cells.
For real-time PCR assays of integrated HIV DNA, cells were snap-frozen after resting cell isolation as above. We thawed and resuspended the dry cell pellets in batches in 200 μL calcium-free and magnesium-free Dulbecco's phosphate buffered saline (Mediatech, Herndon, VA, USA). We extracted DNA with the QIAmp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) and eluted in 50–100 μL of 10 mmol/L Tris, pH 8·0. To isolate total genomic DNA, we electrophoresed up to 2 μg per sample on a 0·5% Seakem Gold agarose gel (Cambrex Bio Science, Rockland, ME, USA) in TAE buffer (40 mmol/L Tris-acetate, 1 mmol/L EDTA, pH 8·3; Invitrogen Life Technologies, Carlsbad, CA, USA) at 60 V for 2·5 h. We excised the band at 23 kbp with a Gene Capsule cutter (Geno Technology, St Louis, MO, USA) and extracted it with a modified QIAEX II protocol (Qiagen, Valencia, CA, USA), shearing the DNA. Briefly, we dissolved gel slices in 600 μL of Buffer QX1 at 50°C for about 15 min. We added 400 μL water and 10 μL QIAEx beads and bound DNA for 20 min at 50°C, mixing beads by inverting tube every 2 min. Beads were pelleted by microcentrifugation at full speed for 30 s. If more than 1 μg DNA was loaded per lane, we subsequently bound the supernatant to a fresh aliquot of 10 μL beads. We washed the beads once with 500 μL Buffer QX1, twice with 500 μL Buffer PE, and dried at 50°C for 10–15 min. We eluted bound DNA with 10 mmol/L Tris, pH 8·0, and we pooled multiple loadings or sequential extractions for a total volume of 60 μL.
We did real-time PCR on an ABI Prism 7700 Sequence Detector in MicroAmp optical 96-well reaction plates with optical caps, using Taqman universal PCR Master Mix and beta actin control reagents from Applied Biosystems (Foster City, CA, USA). We bought NL4-3 gag p24 primers from Integrated DNA Technologies (Coralville, IA, USA. We ran duplicate 50 μL reactions containing 20 μL of sample DNA, 900 nmol/L each PCR primer, and 200 nmol/L probe per reaction with standard curve of 106 to 5 copies of linearised pNL4-3 HIV DNA diluted in 4 mg/L human genomic DNA. Cycle conditions were: 50°C for 2 min, 95°C for 10 min, 45 cycles of 95°C for 15 s, 60°C for 1 min. The amount of DNA assayed was quantified by beta actin real-time PCR with 5 μL of eluate and 2·5 μL of each primer and probe in 25 μL reactions in duplicate. Results are normalised to 500 ng high molecular weight DNA (80,000 resting CD4+ cell equivalents).
Statistical analysis
We estimated the number of resting CD4+ T cells in infected units per billion (109, IUPB) by a maximum likelihood method.28 A parametric, model-based analysis was applied to the preprotocol and postprotocol IUPB data, to quantitatively estimate the effect of enfuvirtide and valproic acid. This approach provides an estimate (95% CI) of the proportional reduction in IUPB after treatment with enfuvirtide and valproic acid, using a generalised linear model with a binary response—ie, a positive or a negative culture.33
Role of the funding source
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
The four patients enrolled tolerated the treatment regimen and adhered well to therapy, according to self-report, refill history, and enfuvirtide vial count (table 1). All had minor reactions at the injection site related to administration of enfuvirtide, as commonly reported.34 Patient 4, with a distant history of antiretroviral-related anaemia and baseline HAART that included zidovudine, developed grade 1 anaemia during the last 4 weeks of treatment with valproic acid. Anaemia resolved when valproic acid was discontinued. Total CD4 and CD8 counts and proportions were stable. With one exception, plasma HIV RNA remained undetectable (<50 copies per mL). Patient 3 had a self-limited upper respiratory tract infection 3–4 weeks after initiation of enfuvirtide, but before initiation of valproic acid. Plasma HIV-1 RNA concentrations at that time were 75 copies per mL, but were less than 50 copies at all other time points.

*Patient 1 had single measurement of 71 copies per mL HIV-1 RNA 96 weeks before initiation of enfuvirtide; patient 3 had single measurements of 636 and 98 copies per mL of HIV-1 RNA 12 and 72 weeks before inititation of enfuvirtide.
&Viraemia suppressed before first visit to our clinic.
Immune activation might have arisen if HIV expression had been induced by valproic acid, and viraemia not contained by HAART and enfuvirtide. We noted no significant changes, however, in concentrations of expression of cell-surface markers of activation at entry (HAART alone), week 4–6 (HAART and enfuvirtide), or week 16–18 (HAART, enfuvirtide, and valproic acid). We detected no significant change in the proportion of resting or activated, memory or naive CD4+ or CD8+ T-cell populations. Lymphoproliferative assay responses to HIV p24 antigen were low at baseline and remained unchanged (data not shown). To assess extent of viraemia, we used a sensitive assay able to detect a single copy of HIV RNA per mL of plasma (table 2).29 Plasma viraemia was undetectable in patient 2. There was no evidence of further suppression of low-level viraemia in patients 1 or 3, nor was there evidence of increased or re-emergent plasma viraemia after addition of valproic acid. Plasma viraemia was initially quantifiable at 1 copy per mL, and then declined to less than 1 copy per mL after addition of enfuvirtide to HAART in patient 4, a decline that might not be significant.



na=not available because of breakdown of real-time device.
*Patient 1=HAART, enfuvirtide, and valproic acid.
†Roche Ultrasensitive plasma HIV-1 RNA=75 copies per mL week 5, <50 copies per mL week 6.
**Patient 1=HAART alone.
^Single-copy assay internal standard below acceptable limit.
Cultures of more than 37 million PBMCs from patient 4 yielded HIV after ex-vivo CD8 depletion and activation at weeks 0–6 (table 2), but none was detected at the end of the study. CD8-depleted cultures did not grow HIV in patients 1 and 2. HIV was also not recovered from CD8-depleted cultures of PBMCs from patient 3, despite low-level plasma viraemia and detectable viraemia at week 5.
We noted a correlation between plasma viraemia and seminal HIV RNA in patients 3 and 4 (semen samples not available for patients 1 and 2). Seminal HIV RNA was not detected at entry, week 6, or week 18 in patient 4 at a limit of detection of less than 200–700 copies per mL. 1025 copies per mL of HIV RNA was detected in patient 3 1 week after low-level plasma viraemia (75 copies per mL) was detected. We did not detect HIV RNA in the semen of this patient at weeks 0 or 18, at a limit of detection of less than 500 copies per mL. We detected integrated HIV DNA genomes much more frequently than replication-competent HIV. During the abbreviated study period, we did not note a substantial decline of integrated HIV DNA. Integrated HIV genomes were detected at week 0 and week 18 in a mean of 275 (SD 88) cells per million resting CD4 cells in patient 1, 375 (288) cells per million in patient 2, 1250 (1013) cells per million in patient 3, and 213 (13) cells per million in patient 4.
The frequency of infection in resting CD4+ T cells on standard HAART was stable (table 3). The greatest decline noted was 28%. Pooled IUPB from resting cell outgrowth assays before addition of enfuvirtide and valproic acid to the treatment regimen was higher than after (table 4). Since we did not recover any virus in outgrowth assays of patient 2 at week 18, maximum IUPB and minimum decline is modelled on an estimate of virus recovery in one macroculture. The decline in resting CD4+ T-cell infection was substantially more rapid (figure) than predicted by the half-life estimates previously reported. Resting cell IUPB declined by at least 29% in all patients. If the IUPB changes after protocol therapy were from the same distribution as the changes in patients on standard HAART, the likelihood that the largest decreases were observed only after treatment with enfuvirtide and valproic acid is less than 0·016 (Wilcoxon rank-sum two-sided test).
Table 3 Frequency of infection in resting CD4+ T cells on standard HAART.
Patient 5 was stably aviraemic as per entry criteria but studied on HAART alone as part of another protocol.
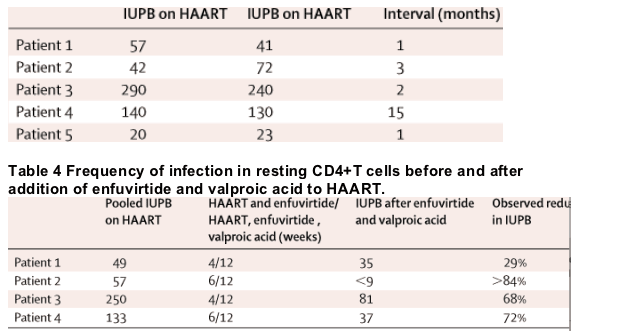
Table 4 Frequency of infection in resting CD4+T cells before and after addition of enfuvirtide and valproic acid to HAART.
We compared the IUPB measured at time points before the addition of enfuvirtide and valproic acid to the IUPB measured after 16–18 weeks of enfuvirtide and valproic acid therapy. In view of the previous estimates of the half life of latently infected resting CD4+ T cells, we tested the null hypothesis that a 50% depletion of IUPB would not be observed after 16–18 weeks of protocol therapy. A decline of more than 50% (95% CI 40–80) was measured in three of four patients after protocol therapy. If the single time point determined is reflective of the decrease in infected resting cell half-life, the half-life of infected resting CD4+ T cells decreased to 2–3 months in the three responders.
Discussion
Our findings show that 16–18 weeks' treatment with a standard clinical dose of valproic acid, in the setting of intensified HAART, produces a substantial decline in the frequency of replication-competent HIV in circulating resting CD4+ T cells.
We measured replication-competent HIV in resting CD4+ T cells under stringent conditions: our volunteers had been successfully treated with HAART for at least 2 years; we used an additional incubation step with reverse transcriptase and integrase inhibitors to reduce to a minimum the likelihood of recovery of replication-competent HIV from recently infected cells; and we did multiple assessments in every patient before initiating treatment with enfuvirtide and valproic acid, none of which showed a consistent fall in IUPB. Depletion of HIV recovered from circulating resting CD4+ cells after treatment, however, indicated a pronounced shortening of the half-life of the latent reservoir.
An examination of the response of individual patients is revealing. Patient 1 had a single episode of transient viraemia, possibly associated with an intercurrent infection, before protocol therapy, and also had low-level viraemia that was unaffected by the addition of enfuvirtide. Resting cell IUPB did not decline much in this patient after therapy with enfuvirtide and valproic acid. Low-level viraemia continued while on intensified HAART. Perhaps these clinical characteristics explain the lack of depletion of resting cell infection seen in this individual. Patient 2 had durable and complete suppression of detectable viraemia, without known episodes of viraemia, without recoverable HIV in CD8-depleted cultures, and without detectable viraemia in the single-copy assay. IUPB of resting cells had fallen greatly by the end of the study. Patient 3 had two episodes of transient viraemia during a state of immune activation before protocol therapy, and one while on study treatment. Low-level viraemia was detected throughout the study, despite the addition of enfuvirtide to HAART. After treatment with enfuvirtide and valproic acid, resting cell IUPB declined. It is noteworthy that resting cell IUPB declined in this patient during therapy with intensified HAART and valproic acid despite continued viraemia. Finally, the IUPB of patient 4′s resting CD4+ T cells measured before protocol therapy was stable. Low-level viraemia was detected before the addition of enfuvirtide, decreased after enfuvirtide therapy, and was less than 1 copy per mL 1 month after enfuvirtide was discontinued. However, this apparent decline might be within the range of experimental variation of the one-copy assay. Although seminal HIV RNA was not detected, HIV was initially recovered from CD8-depleted PBMCs. Consistent with the suppression of low-level replication by intensified HAART, CD8-depleted cultures were negative at the end of study. After 12 weeks of exposure to valproic acid and intensified HAART a decline of resting cell IUPB was noted.
Our pilot study is limited and leaves many questions unanswered. First, we did not note any immune activation in our patients. This finding is probably due to a combination of factors. HIV expressed in resting cells might have been too rare to induce detectable immune activation, a response of HIV-specific T cells present in these durably suppressed patients might have been too rare to detect, or immune responses could have been blunted by effective containment of new rounds of HIV replication by HAART and enfuvirtide. Second, one patient, whose background HAART included zidovudine, developed anaemia. Valproic acid inhibits glucuronidation of zidovudine and increases its bioavailability,35 theoretically increasing the risk of zidovudine-induced anaemia. Whether this adverse effect is infrequently seen in clinical practice, or is under-reported,36 is unclear. In our ongoing studies, we plan to avoid the coadministration of valproic acid and zidovudine. Third, treatment with valproic acid and enfuvirtide seems to have reduced latent HIV infection to an unprecedented extent. We postulate that HDAC inhibition is essential to achieve this effect. However, this study is too small to separate the effects of entry and HDAC inhibition. Furthermore, two of our patients had persistent, low-level viraemia despite the addition of enfuvirtide, an antiretroviral with a new mechanism to which they had not previously been exposed. The source and importance of this viraemia is unclear, and an important topic for future study. Nevertheless, we noted depletion of resting cell infection in one individual despite continuous low-level viraemia. Finally, we have studied a small number of patients, with a single course of therapy of an arbitrary duration. The depletion of resting cell infection we noted seems much greater than that reported after intensification,7 and at least as great as that reported after long-term therapy with interleukin 2.11 However, further studies are needed to confirm, expand, and deepen our observations.
Our findings suggest that eradication of established HIV infection might be achieved in a staged approach. Patients should perhaps first be treated with standard antiretroviral regimens at an early stage of infection. For those in whom viral replication is suppressed, latent viral infection should then be tackled with HDAC inhibitors, intensified therapy, or both. Alternatively, or additionally, therapeutic vaccination might augment the antiviral immune response.
|
|
| |
| |
|
|
|