 |
|
|
| |
Rising Multiple-Dose Assessment of Vicriviroc - Similar Safety, Tolerability, and Pharmacokinetics in Uninfected and HIV-Infected Adults
|
| |
| |
Reported by Jules Levin
A. Sansone1, A. Keung1, M. Caceres1, A. Calzetta1, S. Poggio1, M. Kraan1, R. Rouzier2
1Schering-Plough Research Institute, Kenilworth, NJ, USA; 2Centre CAP, Clinique RECH, Montpellier, France
BACKGROUND
- Vicriviroc is a novel CCR5 receptor antagonist in clinical development for the treatment of human immunodeficiency type 1 (HIV-1) infection. Vicriviroc
works by preventing entry of HIV into T-cells.
- Vicriviroc causes approximately a 1.5 log drop in HIV RNA after 14 days of monotherapy at daily doses of 50 to 100 mg in HIV-1 infected subjects1.
- Vicriviroc has shown synergistic activity with existing antiretrovirals in vitro, and is active against multidrug/ class resistant strains of other classes of
antiretroviral agents2.
- Vicriviroc is rapidly and completely absorbed in rats and monkeys3. Vicriviroc 10 mg administered orally demonstrates good bioavailability and exposure well
above the in vitro IC90 (3.2 ng/mL or 6 nM). Pharmacokinetics of vicriviroc are dose-linear with low to moderate variability in humans4.
- The metabolic pathway for vicriviroc is well characterized in humans; drug interactions are manageable5.
- Vicriviroc is a CYP3A4 substrate6, however it is neither an inhibitor nor an inducer of CYP3A4 orother CYP isoenzymes7.
- Unlike other CCR5 antagonists8,9, vicriviroc is not a substrate of p-Gp10.
Objectives
- The objective of this presentation is to evaluate the safety, tolerability, and pharmacokinetics of vicriviroc in uninfected and HIV-1 infected adults following 14 days of dosing.
- Two placebo-controlled, safety, tolerability, and pharmacokinetics (PK) Phase I studies of vicriviroc at 3 different doses in uninfected and HIV-infected adult subjects were
METHODS
Subjects
Inclusion Criteria
- Healthy men and women based on medical history, physical examination, electrocardiogram (ECG), and routine clinical laboratory tests with and without HIV-1 infection, aged 18- 55 years from two studies
- Body mass index 19- 29 kg/m2
- HIV-1 infected subjects with CD4 cell count 3200, HIV-1 RNA levels between 5,000 and 200,000 copies/mL and no antiretroviral therapy within 8 weeks of enrollment
- Provision of written informed consent
Exclusion Criteria
- Positive for hepatitis C antibody or hepatitis B surface antigen
- Heavy smoking (>10 ten cigarettes or equivalent tobacco use per day)
- Significant food or drug allergies
- Positive drug screen
- History of mental illness or major medical disorders
- History of seizures or head trauma with loss of consciousness
Study Design
- These two studies were randomized, third-party blind (within dose level), placebo-controlled, rising multiple-dose studies.
- Subjects were randomized sequentially to vicriviroc 10 mg, 25 mg, or 50 mg twice daily (BID), or matching placebo for 14 days in a ratio of 3:1.
Treatment Regimen
Subjects were enrolled into 1 of the following treatments and then
randomized to vicriviroc or placebo:
- Treatment A: Vicriviroc 10 mg or placebo administered BID for 14 days
- Treatment B: Vicriviroc 25 mg or placebo administered BID for 14 days
- Treatment C: Vicriviroc 50 mg or placebo administered BID for 14 days
Assessments
- Safety and tolerability assessments were based on occurrences of adverse events (AEs), vital sign measurements, physical examinations, ECG results, and clinical laboratory values.
- To determine the pharmacokinetics of vicriviroc in uninfected and HIV-1 infected subjects, blood samples were drawn:
- On Day 14, blood samples were collected at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72 (last sample collected for uninfected subjects), and 96 hr (last sample collected for HIV-1 infected subjects) after the AM dose.
- All plasma samples were assayed using a validated highperformance liquid chromatography method with tandem mass spectrometric detection.
- Primary PK parameters were maximum measured plasma concentration (Cmax) and area under the plasma concentration-time curve after dosing (AUC), time to Cmax (Tmax), trough plasma concentration (Cmin), terminal phase half-life (t1/2), apparent volume of distribution (Vd/F), and total body clearance (CL/F).
Subject Disposition and Demographics
- A total of 37 healthy adult uninfected subjects were enrolled (36 males, 1 female) between the ages of 18 and 47. Thirty-five subjects were caucasian and 2 were black.
- A total of 49 healthy adult subjects with HIV-1 infection were enrolled (40 males, 9 female) between the ages of 25 and 53. Forty-three subjects were caucasian, 3 were black, and 3 were hispanic.
Safety Assessment
- Twenty-one percent (6/28) of uninfected subjects receiving vicriviroc compared to 22% (2/9) of uninfected subjects receiving placebo reported 31 AE (all mild to moderate, none considered treatment related). No HIV-uninfected subjects were discontinued due to an AE.
- Seventy-two percent (26/36) of HIV-infected subjects receiving vivriviroc compared to 62% (8/13) of subjects of HIV-infected subjects receiving placebo reported 31 AE (most common after vicriviroc: headache, 28%; abdominal pain,
14%; nausea, 14%). No AEs were dose related, 3 AEs were considered serious.
- One SAE occurred before dosing.
- Two SAEs were incurrent infections with a possible relationship to vicriviroc.
- In the HIV-infected study there were 3 serious AEs
- In both studies, none of the AEs were dose related and clinical laboratory tests, vital signs, and ECG revealed no remarkable findings.
Pharmacokinetic Analysis
- Mean plasma vicriviroc concentration-time profiles following oral administration of vicriviroc to subjects with or without HIV-1 infections are presented in Figure 1.
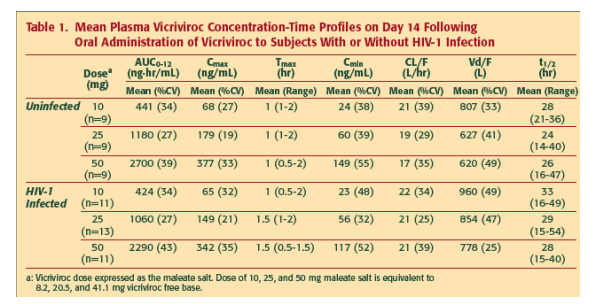
- At the lowest dose (10 mg) vicriviroc demonstrates good bioavailability, and exposures are well above the in vitro IC90 (3.2 ng/mL or 6 nM) on Day 14 throughout the dosing interval (Table 1).
- Exposure increased in a dose-linear manner with low-to-moderate inter-subject variability (%CV ~20-40% for Cmax and AUC) in PK (Figures 2 and 3, Table 1); steady state was attained by Day 14 for all doses.
- Vd/F of vicriviroc was large (mean Vd/F ranged from 620 to 960 L), suggesting extensive distribution into extravascular tissues (Table 1).
- Both mean CL/F and terminal phase t1/2 values were similar between studies and t1/2 ranged from 17 to 22 L/hr and 24 hr to 33 hr, respectively, indicating vicriviroc was cleared slowly from the body (Table 1).
CONCLUSIONS
- Vicriviroc was well tolerated in both HIV-1 infected and HIV-1 uninfected subjects.
- The results indicate similar pharmacokinetics between subjects with or without HIV-1 infection.
- Vicriviroc was rapidly absorbed, had a long half-life, good bioavailability, and similar dose-linear exposure at 20, 50, and 100 mg daily in uninfected or HIV-infected subjects.
Therefore, data from healthy volunteers is a reliable predictor of what will happen in the HIV infected treatment population.
|
|
| |
| |
|
|
|
|
|