 |
|
|
| |
Antiretrovirals at
Denver: 13th CROI 2006 - "Future Seems Bright"
|
| |
| |
Written for NATAP by Mike Youle, MD, Royal Free Hospital, London, UK
Like a kid in a candy store, there was so much new data at this meeting it was hard to concentrate and the possibilities now for our patients are immense. Notwithstanding the early stage of development of most of the new agents shown, the future seems bright. After the disappointment of the recent loss of one or maybe out of three CCR5 inhibitors in clinical studies the integrase inhibitors from Merck and Gilead burst onto the scene and the latter looked even better than the first set of data shown at the Dublin EACS conference last November. In addition there were many new agents in the wings such as the once weekly fusion inhibitors from Trimeris and further information on those drugs closest to availability, TMC114 and TMC125. This was all good new since the venue city reminded me of Brokeback Mountain meets Eraserhead, not a place I wish to return to quickly.
So here is the selection of new drug data presented at the meeting:
In the main body of the report I will focus those agents that have clinical data or are nearer to development. However, in earlier stages of development are some compounds which may yield useful classes of drugs in the future. There was a study presented by Donahue and co-workers from Vanderbilt which screened peptide inhibitors of the binding of HIV-vif to human APOBEC3G [200]. HIV-vif inactivates a host cell antiviral defence mechanism mediated by the cytidine deaminase APOBEC3G and thus inhibition of this interaction may be a disadvantage to viral replication. A novel innate protein, G9, blocks the HIV life-cycle at two stages: it inhibits HIV production prior to release of virus particles and inhibits an entry -dependent step at reverse transcription. Findings by a group at King's College London suggest this to be a potential immunotherapeutic [210].
Several studies furthered the knowledge base on short interfering RNA's (siRNA's) by evaluating the utility of targeting the U3 region of the 3'LTR which is utilised in all viral transcription [266]; and in the second study showing that for the full effect of an siRNA full homology (complete matching) is required with the target sequence of the gene [267]. A third study from Pauls and co-workers in Badalona suggest that it is important to develop siRNA sequences that are devoid of unwanted inflammatory activity that may increase rather than reduce HIV activity [514]. Although this area of research is clearly very complex the goal of safe targeted silencing of HIV-replication with all cells is one worth fighting for.
Robert Gallo and his group in Maryland investigated indirubin 3'-monoxime, a derivative of a Chinese medicine used to treat leukaemia and showed that in non-toxic concentrations that did not affect cell proliferation the compound can reduce tat-mediated transactivation an obligate step in the production on HIV-1; the drug is effective against wild-type and resistant strains of HIV [510].
Thomas Duensing from Tanox Inc showed new data on the in vitro characteristics of HIV isolated from patients treated with the novel entry inhibitor TNX-355 [158LB]. TNX-355 is a monoclonal antibody that targets conformational changes in gp120 and data was presented on ongoing phase II study, first shown at ICAAC 2005. In this presentation they showed the first data on resistance to TNX-355; IC50 was 10-230ng/mL and there appeared to be no effect due to co-receptor tropism. From baseline to week 9 three of 19 subjects remained fully susceptible to the agent whilst the other sixteen had reduced activity by 19-60%. Sub-clones were isolated and grouped into sensitive 90%, intermediate 42% and resistant 17%. Using soluble CD4 it appears that there is an increased risk of neutralisation when there is increased exposure to the CD4 binding site in TNX-355 resistant env proteins. Further studies are ongoing to correlate baseline susceptibility with virological outcomes.
There was a little more information pertaining to maraviroc (formerly UK427,857), the only CCR5 currently left in the running at the level of clinical trials, although Schering-Plough may enter the race once again with higher doses of vicriviroc. The group from Pfizer Global R&D, based in Sandwich in the UK, examined the activity of the agent on human CCR5 and related this to its' binding characteristics [504]. They showed that against another CCR5 blocking compound maraviroc exhibited marked viral suppression commensurate with a long dissociation half life of 15.95hours, suggesting that the better activity was due in a major part to the prolonged drug binding to the receptor. Resistance work performed by this unit in collaboration with Monogram Biosciences characterised resistant NL4-3 clones which resulted in dose response curves that did not produce 100% inhibition of CCR5 receptor potentially due to the selection of viral variants which recognise receptor: inhibitor complexes [598].
The pharmacokinetics of CCR5 antagonist vicriviroc (VVC) in combination with ritonavir were assessed in cohorts of 8 healthy volunteers given atazanavir, indinavir, nelfinavir, saquinavir or fosamprenavir [582]. There were no significant changes in plasma concentrations when combined with any of these agents, irrespective of p-glycoprotein or CYP450 interactions.
Wayne Greaves from Schering-Plough presented study P03802 (design shown below) a phase 2 randomised placebo controlled study of vicriviroc in treatment naive patients with R5- tropic virus which was terminated early by the Data and Safety Monitoring Board in October 2005 [161LB].

In hind-sight it appears as if doses of vicriviroc may have been too low. The relative rates of virologic breakthroughs in the vicriviroc arms compared to the placebo arm were 56% (13/23) in the 25 mg dose arm (vs control, p<0.001), 41% (9/22) in the 50 mg arm (vs control, p=0.003), and 17% (4/23) experienced viral breakthrough in the 75 mg (vs control, p=0.188). At the time of study termination, there was no significance difference in viral failure breakthrough between the 75 mg arm & the EFV/CBV arm.
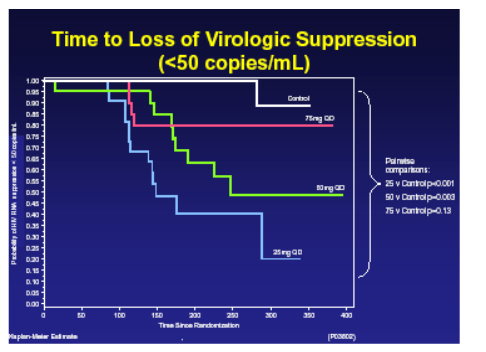
22 of 26 of breakthroughs on vicriviroc had evaluable genotypes and of these 100% had M184V and one had M184V and M41L, resistance to the CCR5 inhibitor is still being investigated, no correlation was seen with IC50 and no consistent mutations in envelope arose. There were no safety concerns and specifically no liver function abnormalities were seen. The assessment of a more appropriate dose will now take place to see if this compound has a future.
Down regulation of CCR5 expression on monocyte derived macrophages was achieved using aprepitant (an FDA approved anti-emetic which works by antagonising neurokinin-1 receptors) in a study by Wang in the Children's Hospital of Philadelphia [511]. It works on AZT resistant virus as well as inhibitor of R5 but not X4 viruses but seemed not to work in all donor isolates.
There were two presentations of human monoclonal antibodies targeted at CCR5. PRO140, a humanised IG4 mAb from Progenics was given intravenously to healthy male volunteers as single doses of 0.1, 0.5, 1.0 and 5.0mg/kg in sequential dose rising cohorts of 4:1 drug: placebo [515]. All 20 subjects finished the study with no reported side effects of ECG changes and the serum concentrations increased proportionally with dose and decreased with a serum half-life of around two weeks. At the 5mg/kg dose there was coating of CCR5 lymphocytes for >60days suggesting that this agent although parenteral may be able to be dosed quite infrequently. A second mAb, in this case from Human Genome Sciences, mAb004 was examined in vitro and showed little tendency to result in resistance over at least 24 weeks [505].
Another monoclonal antibody approach was taken by Yoshimura and co-workers from Kumamoto University in Japan when they synthesized a mAb, KD-247 focussed on the highly conserved V3 sequences and which showed a high degree of synergism with available CCR5 inhibitors [506]. At high concentrations of 1000_g/mL it was possible to select a mutation, G314E, in the tip of the V3 loop, which conferred complete resistance to the compound but rendered it hypersensitive to several other agents.
Finally for CCR5 targeting a novel approach was reported by Elena Perez from Sangamo Biosciences where they had created a zinc finger protein nuclease (ZFN) to target the CCR5 gene to create a double stranded break (DSB) at predetermined sequences [51] which then are repaired by host DNA repair mechanisms but the target gene is permanently disrupted. These ZFN can target any sequence in the genome and produce a break which will disrupt transcription and the group picked CCR5 as a suitable target. They showed that a cell line modified by ZFN-targeting of CCR5 was resistant to HIV infection but that this could be reversed by the restoration of CCR5 by plasmid nucelofection. The group are currently trying to optimise this approach.
In terms of CXCR4 blockade an oral presentation of two new compounds, KRH-3955 and KRH-3140 was presented by Yuetsu Tanaka [49LB]. These selectively block binding of SDF-1_ and were administered to rats and showed good PK with levels above the EC90 for 23 hours; no toxicity was seen, other than a mild rise in white blood cells. KRK-3955 was present two weeks after a single oral dose of 10mg/kg and was protective to a high degree against an X4 virus challenge.
The last group of agents to be show-cased at the conference that act on the entry mechanism of HIV into the cell were very exciting. The joint-venture between Trimeris and Roche has already produced enfurvitide (T20) which in the TORO studies showed a marked benefit compared to the optimized background alone and in further studies of newer drugs such as tipranavir (RESIST) and TMC114 (POWER) has marked itself out as the best companion agent for these in three-class failing patients. However with its' twice daily injections, injection site reactions and mismatch between patient wishes and physician willingness to offer and prescribe the drug has not been used optimally to date. However after an aborted attempt to improve the agent with T1249 there now appears to be the possibility of once weekly dosing with new lead compounds TR290999 and TR29144 [48]. In an excellent presentation by Barbara Delmedico, the Jody Foster of Trimeris, we were given an overview of these exciting new generation fusion inhibitors (NGFI) which not only have pharmacokinetic properties that allow for once weekly dosing but also appear to be significantly more potent against HIV and also to have activity against enfurvitide -resistant strains; I was on the edge of my seat throughout!
They developed a lead compound, T 651, which spanned more of the HR2 domain of gp41 and represented an optimal slice of this with which to bind and disrupt the process of fusion. As you see below this agent had a much greater activity than T20 but unfortunately did not have good pharmacokinetic properties.
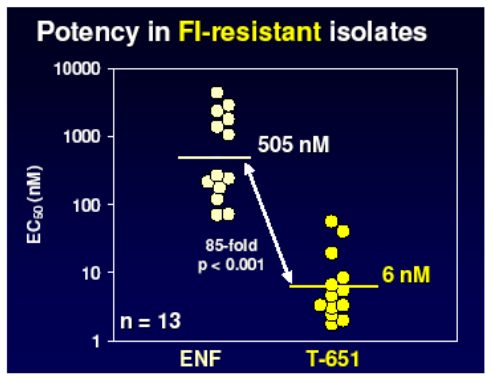
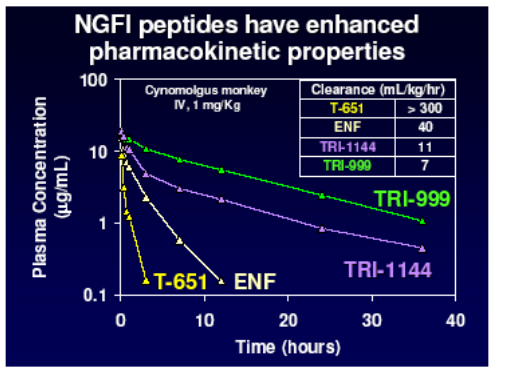
However further work produced two other chemical series with enhanced properties and much improved PK raising the possibility of once weekly dosing by the addition of helix stabilisation motifs (TRI-1144 series) or fatty acid acylation (TRI-999 series).
Lead compounds from these were then evaluated against a panel of viruses resistant to enfurvitide and showed between 150 and 250 fold improvement over the current compound.
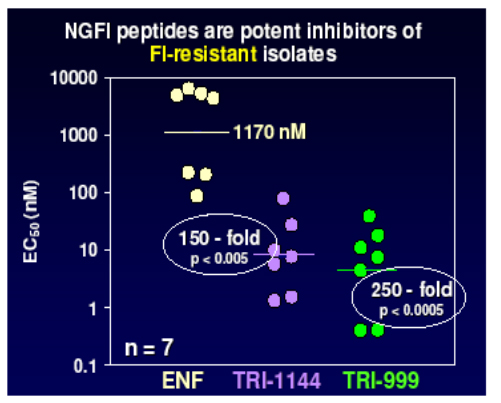
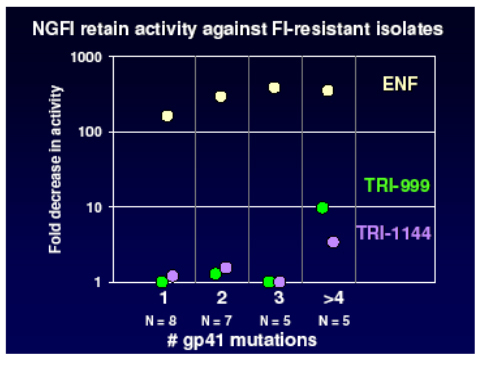
In addition the NGFI retain sensitivity against mutants with up to 4 codon changes in gp41 as shown in the panel below, whilst enfurvitide loses activity after the acquisition of only one mutation. This bodes well for the development of once weekly agents with a high genetic barrier which will be useful both in patients naive to enfurvitide and in those who have acquired resistance.
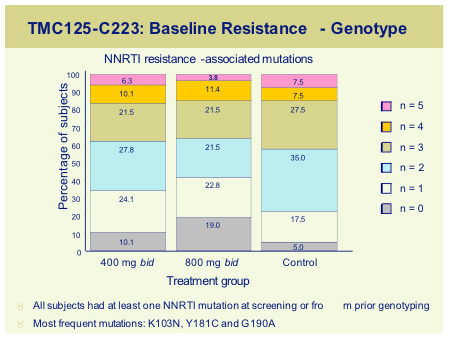
Non-nucleoside reverse transcriptase inhibitor (NNRTI) data at the conference was focussed on the Tibotec compound, TMC125. TMC125-C223, was a randomised dose finding study, run in the USA in 199 subjects with documented resistance to NNRTI's and >3 PI primary mutations [154]. Subjects either received TMC125 (400mg or 800mg) with an optimised background, or optimized regimen alone. Median VL at baseline was 4.7log and T4 around100 cells and baseline genotypic resistance is shown below.
For the 800mg dose no single mutation was associated with a >10 fold reduction in sensitivity, whilst the following mutation, always in combination with up to 4 other mutations produced a >10FC; K10P, V179E, V179F, Y181C, Y181V, G190S, M230L, all previously found in vitro to be associated with an increased FC to TMC125.
The viral load reduction was around 1 log but number of baseline NNRTI mutation predicted the change in viral load with a -0.66 log reduction even with some 3 NNRTI mutants, compared to only -0.19 for the control subjects.
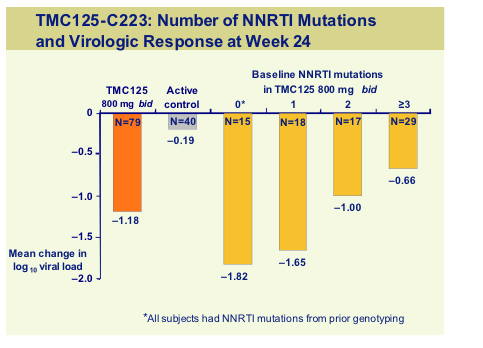
This is a 48-week study so further data will be available for the target dose of 800mg bid but at this mid-point analysis. TMC125 retained activity in the presence of multiple NNRTI mutations. However as with all new agents it is vital that this drug be used in the setting of an active background and data suggests that this drug can be used successfully with TMC114 [575c].
A PK study of the combination of TMC125 800mg bid and tipranavir/r 500mg/100mg was conducted as an open-label cross-over randomised trial in 24 healthy volunteers [583], the AUC was 24% (CI:18-33%;p<0.05) compared to the administration of TMC125 alone. Cmax and Cmin were 29% and 18% respectively whilst tipranavir levels were unaffected by the combination. Thus this combination of agents does not appear viable due to the marked reduction in TMC125 levels. A second PK study of subjects already taking saquinavir/lopinavir/ritonavir (800/400/100mg bid) plus NRTI's was conducted in patients in Canada by Marianne Harris [575b]. All subjects had previous NNRTI resistance and viral loads <50 when studied; the combination seemed safe and no rashes or cardiac events were seen. Cmax and AUC12h for lopinavir were significantly reduced by 24% and 19% respectively (p=0.03 and P=0.04) but were not thought by the authors to be clinically significant, no change in PK were seen for the saquinavir, ritonavir or TMC125.
Gilead Sciences has emerged as one of the most innovative drug companies in the HIV-field and their new nucleotide reverse transcriptase inhibitor is G9148 active as an active diphosphate metabolite. The compound uses amidate pro-drug technology to maximise the intracellular levels of active compound and in this study in Beagles the drug accumulated substantially within PBMCs [498] and has low mitochondrial toxicity in renal, bone marrow and liver cell lines.
Tomas Cihlar presented in vitro data on the drug tested against a wide variety of NRTI resistant strains [45]. It showed that activity of GS9148 was unaffected (FC<1) by K65R, L74V, M184V or any of these in combination as well as retained sensitivity to up to 4 TAMS (FC<2) making this drug an attractive candidate for further studies.
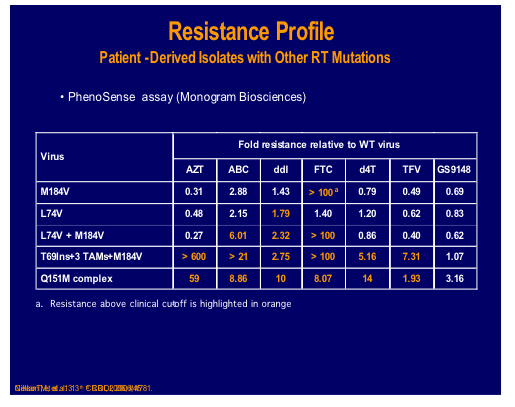
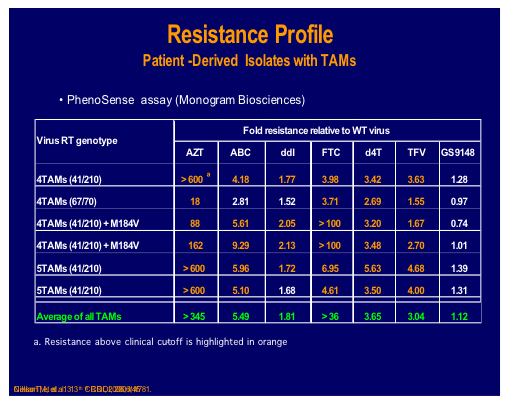
A nucleotide-competing reverse transcriptase inhibitor (NcRTI-1) was showcased by Matthias Götte from McGill University, Montreal [47]. This agent blocks DNA polymerase and appears to occupy the nucleotide biding site of HIV-1 RT and forms a dead-end complex that prevents the incorporation of dNTP substrates. This prototype compound is active against NNRTI resistant viruses but is adversely affected by M184V (fold change reduced by 5-fold), which is clearly a problem for this particular compound whereas K65R confers hyper susceptibility.
NcRTI -1 was examined for its' sensitivity to isolates and site directed mutants containing many different NRTI mutations and showed 39 fold decreased susceptibility to the combination Q151M and M184V, although thymidine analogue mutations (TAMs), 69 insertion or Q151M alone showed no decrease [500].
A potent thymidine kinase dependent with low toxicity would be useful to replace zidovudine and stavudine and dioxolane thymidine (DOT) was presented as a candidate by John Lennerstrand of Emory University [46]. It seemed to work well against most NRTI mutations other than Q151M but the presenter seemed reticent to clearly answer questions regarding pharmacokinetics.
Further NRTI titbits included an in vitro study of E2fDA, which has a long intracellular half-life, minimal inhibition of DNA polymerase-_ and good activity against resistant strains [499]. Dexelvucitabine (formerly Reverset) resistance was assessed in a study whose clinical data was presented in Rio last summer [632]. Apart from reduced sensitivity to Q151M, no other mutations were associated with baseline phenotypic resistance or poor virological outcomes over 16 weeks of therapy.
Integrase Inhibitors Brand NEW, Highlight of the Conference
MK-0518, the new integrase inhibitor from Merck, is an agent that prevents HIV from incorporating its genetic information into that of the host cell and thereby prevents established infection of T cells. It has shown good efficacy in vitro and is synergistic with many available HIV drugs and since it has a novel mode of action, within the nucleus of the cell, there should be no cross resistance and thus represents a new option for those with multi-drug resistance. The good news continued with no safety issues from early studies and good tolerability and no requirement to dose with food. The update on this drug following its' first showing at the Dublin EACS was given by Beatriz Grinsztejn of the Oswaldo Cruz Foundation in Rio de Janeiro [159LB].
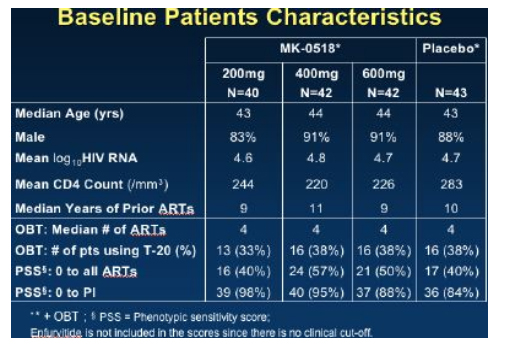
Protocol 005 was a randomized, double blind study of MK-0518, 200, or 400, or 600 mg MK-5018 bid, oral administration vs placebo: all in combination with optimized background therapy (OBT). Baseline stratification was performed for use of enfurvitide in the OBT (about 30% used T20) and level of resistance to PI's at entry. In addition there was a sub-study comparison made between those who included atazanavir or not in the OBT since this agent; atazanavir is an inhibitor of UGT1A, which could have potentially affected the levels of MK-0518 (but did not appear to do so significantly). Subjects were 3 class experienced with an HIV RNA >5000 and CD4 >50 cells. Baseline characteristics of the participants are shown below.
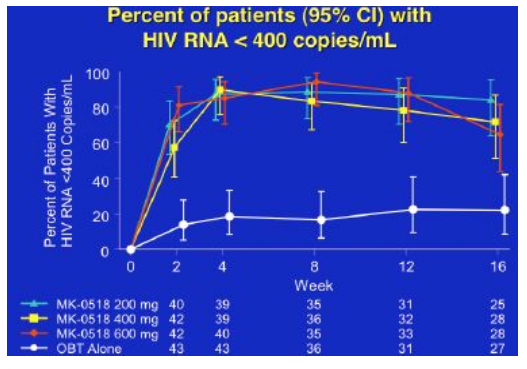
By week 16 with 25-28 patients in each group, 65% to 85% of patients in the MK-0518 dosing arms had HIV RNA <400 and 56% to 72% of patients in the three MK-0518 dose groups had <50, compared to 20% in the placebo (OBT) group.
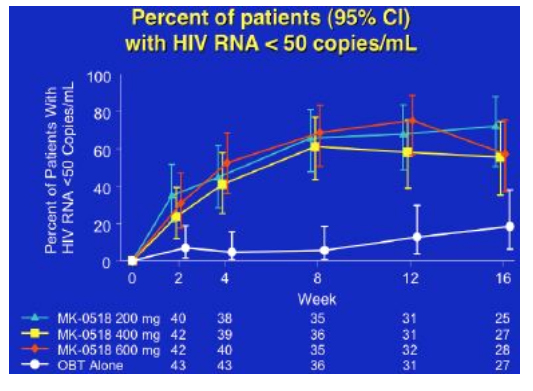
Over 16 weeks CD4 rise was around 100cells for the 400 and 600mg dose groups, and about 30cells for the 200mg dose group and the placebo group, whilst the mean viral load decline was approximately 2logs for the MK-0518 groups compared to just under 1 log for the comparator arms, although since many subjects were undetectable the absolute benefit of MK-0518 may be greater.
Few adverse events were reported and the MK-0518 arms seemed to be similar to the placebo group with diarrhoea, nausea and fatigue being the commonest problems, grade 3/4 laboratory abnormalities were also similar between MK-0518 arms and placebo.
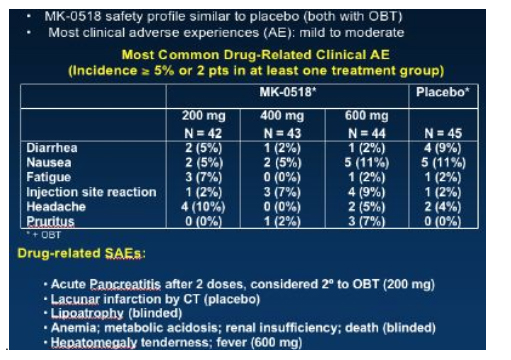
So overall an extremely exciting presentation of this new drug which hopefully will be available on expanded access in the last quarter of 2006.
The second company to enter the Integrase arena is Gilead with a drug developed originally by Japan Tobacco (JT-303/GS9137), in vitro data shows this compound to have an IC50 of around 8.8nm and is 3-fold more potent than the diketo acid L-870810, originally studied by Merck [508]. It retained full sensitivity to eight sub-types and a panel of resistant viruses and showed synergy to 3TC as well as additive to most other antiretrovirals. Healthy volunteers were dosed with 100-800mg as single doses in a dose-escalation study (6 active drug and 2 placebo subjects per group) [580]. No tolerability issues were seen, plasma concentrations exceeded the protein binding adjusted EC90 at 24 hours and the administration of drug with food produced a 3-fold increase in Cmax and AUC.
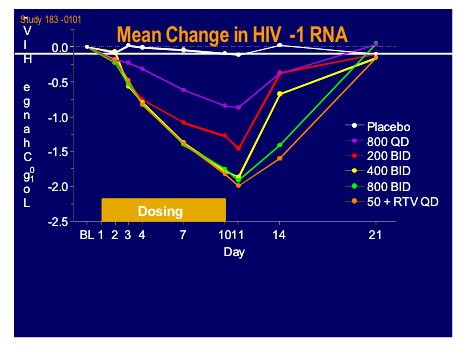
Edwin deJesus from Orlando gave a presentation of the latest clinical data from the 183-101 study which was a randomised sequential dose cohort, double-blind, placebo controlled monotherapy in 40 patients, of whom 15 were naive (11 active) and 25 treatment experienced currently not on therapy (19 active) with viral load between 10,000 and 300,000 subjects and a CD4 count > 200cells (mean VL 4.75log, T442) [160LB].
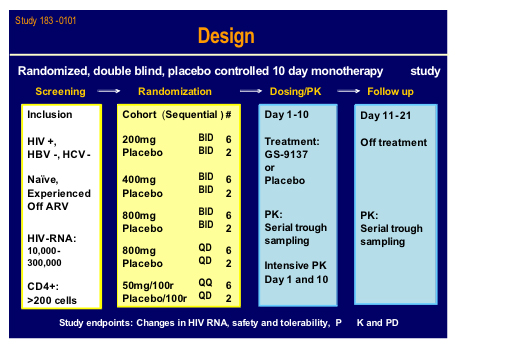
Ritonavir boosting clearly helped the PK and may give the option for once daily dosing.
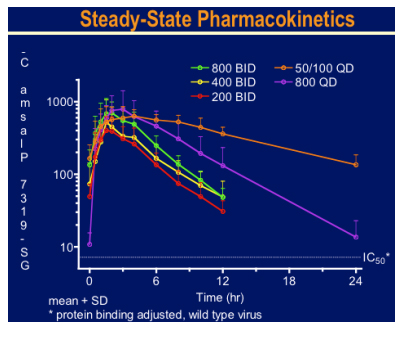
There was a brisk drop in viral load over the 10 day treatment period to around 2log with 800mg bid and 50mg plus 100mg ritonavir qd arms. Side effects were minimal and no specific drug related toxicity was seen. This looks an extremely exciting new agent.
Two new protease inhibitors had their first showing at this meeting. SPI-256 developed by Sequoia Pharmaceuticals was assessed in vitro against a panel of resistant viruses by Monogram Biosciences [501]. In a subset of isolates with >50 fold resistance to current licensed PI's and at least 6 primary mutations average IC50 values to SP-256 were12.9nM compared to >937nm for the reference agents, giving a-50 fold increase in efficacy. The second new kid on the block was a novel non-peptidic PI, GRL-02031 which although tested against less resistant strains than the former compound seemed to be sensitive to many currently resistant isolates [503]. It is comforting that there still seems to be a pipeline of new agents in this class.
More data was shown on TMC114 including a pharmacokinetic (PK) study to assess the effect this drug on the levels achieved when given in combination with TMC125 by Marta Boffito from London [575c]. Kim Strubble from the FDA, in here talk on drug-drug interactions [53] highlighted the difficulties inherent in PK studies in highly experienced patients and notes that the use of historical control data in stead of monotherapy arms may be permitted. This combination of TMC114 600mg/ritonavir100mg/TMC125 200mg twice daily was given to 10 subjects with multi-drug resistance as a part of a salvage regimen. The median CD4 at baseline was 75 and viral load 4.6log. All patients had a 2 log drop by week 6 (median -2.55log) and 8/10 achieved a viral load <400. As compared to historical controls exposure to TMC114 was unchanged and that to TMC 125 modestly reduced by 30%, a change deemed non-significant. A second PK study examined the PK/pharmacodynamic analyses of TMC114 in the Power studies and found that the outcomes were strongly influenced by baseline TMC114 fold change, viral load and use of sensitive drugs in the optimised background [639b]. However the strongest predictor of response was the sensitivity to TMC114.
In an oral presentation by Marie-Pierre de Bethune from Tibotec this relationship was examined further. The drug has shown good responses compared to the comparator PI's even if there was retained susceptibility to those drugs.
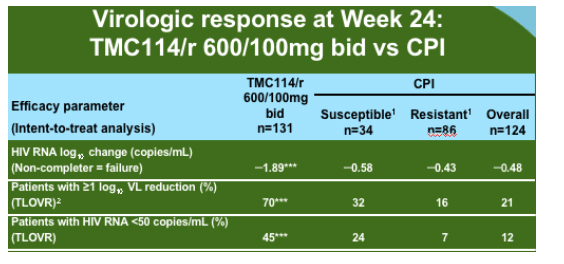
The group analysed all of the data available for the 600mg/100mg dose from the two presented Power studies and additional information form the Power 3 study which has the same entry criteria and has increased the sample size of the original studies to 458 of which 377 have reached week 24 or have discontinued. As compared to the comparator PI background TMC114 was relatively unaffected by PI mutations at baseline until there were 12 or more.
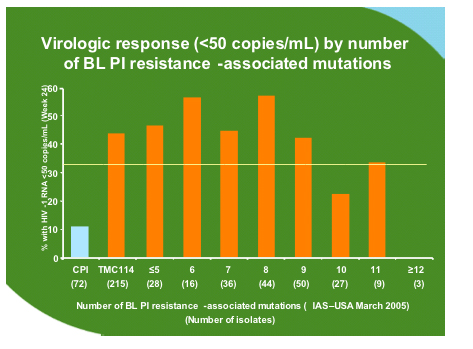
No particular mutation was associated with failure but some mutations developed commonly during treatment with TMC114 as shown below. However when these were inserted as codon changes in site directed mutants along with 1-2 other mutations this did not affect sensitivity to TMC114.

In rebounders the FC for TMC114 increased by a median of 8.14 from baseline but that for tipranavir there was only a median fold change of 0.82 from baseline suggesting that cross-resistance between these two drugs may not be an issue.
There were three presentations on PA-457 the derivative of betulinic acid and the first in class maturation inhibitor. In vitro data where the agent was tested against a panel of site directed mutants showed no resistance and either an additive or synergistic effect with all available antiretroviral classes [509]. In a PK/PD study 32 subjects were given oral liquid doses of 25, 50, 10, 200mg once daily for 10 days as, monotherapy, after a one day double loading dose (6 received active drug and 2 placebo in each dose group) [52]. PA-457 was well tolerated, had a half life of 72.8 hours and gave up to a 1.1log drop at the highest dose level. Using an E-max model to assess optimal dose it seems that the dose can be increased substantially. There were two subjects that did not show any response and this did not correlate with drug exposure. The final talk on this drug, by Catherine Adamson from the HIV Drug Resistance Program at NCI examined the resistance patterns in vitro by serial passage and from gag sequences selected from the phase I study above [156]. She gave a very clear description of two groups of identified mutations located at the C-terminus of the capsid protein and in the first and third residues of the spacer peptide SP1. The clustering of these PA-457 resistance mutations at the CA/SP1 junction confirms this as the site of action of the drug. No resistance was seen in the clinical isolates.
Finally in the early stages of evaluation is a compound by Pfizer, UK201844, found by high throughput screening at the rate of 72,000 compounds per week, which appears to be a maturation inhibitor focussed on gp160 processing, inhibiting the incorporation of functional HIV-env into virions [50LB]. The lead compound does not have overall strain inhibition (only 1/15 clade B isolates) and further work will be needed to ascertain how active agents may be created.
And the postscript of course is that I am writing this report snowed in at Newark Airport waiting for my beleaguered British Airways plane to have 26 inches of snow swept off it so I can get back to my clinic!
Thank you for those investigators who shared slides from their presentations.
Mike Youle
|
|
| |
| |
|
|
|
|
|