 |
|
|
| |
Antilipid Tactics, Diabetes, and Abacavir Reactions:
(1) prescribe a lipid lowering drug or switch to NNRTI; (2) Atazanavir, lipids, and cardiovascular risk; (3) antilipid drug rosuvastatin; (4) Diabetes risk factors in D:A:D: d4T, ddI, AZT increase risk, nevirapine protective; (5) abacavir genetic testing prevents hypersensitivity reaction
|
| |
| |
Glasgow Meeting: Part 2
8th International Congress on Drug Therapy in HIV Infection
November 12-16, 2006, Glasgow
Mark Mascolini
What's the better way to rein in runaway lipids: switch to a nonnucleoside (NNRTI) or use an antilipid drug? The 8th Glasgow meeting showcased a big analysis of the D:A:D cohort that offered some answers to that question. Other Glasgow studies looked at how different ritonavir-boosted protease inhibitors (PIs) affect not only lipids, but also endothelial function and cardiovascular risk. Another D:A:D inquest probed for diabetes risk factors in people with HIV. And an interesting British study sized up routine HLA screening as a way to predict--and maybe prevent--abacavir hypersensitivity reactions.
Prescribe a lipid lowerer or switch to a nonnuke?
A year ago Italian investigators published results of a 130-person randomized trial showing that a fibrate or statin deflates total cholesterol better than swapping a PI for efavirenz or nevirapine after 12 months of follow-up [1]. Researchers from the 11-cohort D:A:D group mimicked parameters of that study to see if the results held up in a larger group of patients [2].
D:A:D's data banks now embrace 33,389 HIV-infected people in Europe, Australia, and the US. Marc van der Valk (Academic Medical Center, Amsterdam) and coworkers picked out everyone who had taken a PI for more than 6 months, had never taken an NNRTI, and had a total cholesterol above 6 mmol/L (230 mg/dL) twice in a row. They divided them into three groups--a lipid-lowering group of 221 people who started a fibrate or statin, a switch group of 208 people who traded a PI for an NNRTI, and a 1463-person control group that made no changes in antiretroviral therapy and tried no lipid-constraining strategies. Through 12 months of follow-up, the study design allowed no further changes in antiretrovirals in any group or any additional antilipid maneuvers.
The antilipid and switch groups were significantly older than controls (45 versus 41 years, P = 0.0001) and included more men (86% versus 82% of switchers versus 79% of controls, P = 0.08). More antilipid takers had a history of heart disease (4.5% versus 0% versus 1.2%, P = 0.0005), took drugs for heart disease (15% versus 8% versus 7%, P = 0.0004), and had diabetes (10% versus 4% versus 2%, P = 0.0001). The antilipid contingent also took ritonavir with or without lopinavir more often than other groups. Switchers had higher CD4 counts (534) than the antilipid group (484) or the control group (488) (P = 0.0003). Baseline total cholesterol, dangerous low-density lipoprotein (LDL) cholesterol, total-to-high-density lipoprotein (HDL) ratio, and triglycerides were all significantly higher in the antilipid group than in the other two groups, while "good" HDL cholesterol was significantly higher in the control group. The three groups did not differ in body mass index or smoking history.
After a year of follow-up total cholesterol fell about 1 mmol/L in the antilipid group (P < 0.0001 versus controls, see note 3) and about 0.6 mmol/L in the switch group (P = 0.02 versus controls) (P = 0.0001 for all three groups versus baseline). "Good" HDL cholesterol rose about 0.18 mmol/L in the switch group (P = 0.0001 versus controls) and about 0.07 mmol/L in the antilipid group (a nonsignificant difference versus controls or baseline). Total-to-HDL cholesterol ratio fell about 1.5 in both the switch and antilipid groups (P = 0.0001 versus baseline for both, P = 0.06 for antilipids versus controls, P = 0.03 for switch versus controls).
Ominous LDL cholesterol fell most in the antilipid group (about 0.9 mmol/L, P = 0.0004 versus controls), while dropping about 0.7 mmol/L in the switch group (P = 0.06 versus controls) and 0.5 mmol/L in the control group (P = 0.0001 for all group changes from baseline). Triglycerides, on the other hand, fell most in the switch group (about 1.0 mmol/L, P = 0.0001 versus controls), while tumbling 0.8 mmol/L in the antilipid group (P = 0.007 versus control).
In short, lipid-lowering therapy yielded the biggest declines in total and LDL cholesterol, particularly in people with the highest baseline levels of these lipids. Those results confirmed the Italian trial findings [1]. But the D:A:D team judged antilipids' effects on HDL cholesterol "minimal." Swapping a PI for an NNRTI did best in pumping up HDL cholesterol, regardless of baseline level. The two tactics had a similar impact on total-to-HDL cholesterol ratio.
This analysis has limits, van der Valk warned. Because cohort members were not randomized to a lipid-controlling strategy, selection bias could affect the results. By excluding people who tried switching to an NNRTI then started antilipid drugs, the study may have excluded a set of patients with hard-to-control hyperlipidemia. Baseline lipids varied markedly from group to group. And D:A:D researchers had no information on possible lifestyle changes, specific antilipid drugs or doses, or whether lipid samples were drawn after fasting.
Retrospective look at rosuvastatin for unruly lipids
Clinicians from three British hospitals reported good early control of total cholesterol and triglycerides with rosuvastatin in 33 HIV-infected people, 32 of them taking antiretrovirals [4]. Rosuvastatin merits attention as an antilipid agent because its metabolism does not depend on liver enzymes involved in metabolizing PIs or NNRTIs.
The study group had a median age of 46 years (range 32 to 66), 26 (78%) were white, and 19 (57.5%) were gay. They began the statin at a 10-mg once-daily dose, which rose to a maximum of 40 mg in 2 people. Eight people also took a fibrate, 2 took omega-3 fatty acids, and 1 took ezetimibe. Of the 32 people taking antiretrovirals, 21 (66%) were taking an NNRTI and 11 (34%) a boosted PI for a median of 52 months (range 10 to 104).
Satyajit Das (University Hospital Coventry and Warwickshire) reported that average total cholesterol and triglycerides fell significantly in the first 3 months of rosuvastatin therapy, but not in later months (Table 1). HDL cholesterol did not rise significantly. Drops in total cholesterol and triglycerides correlated with higher baseline readings (r = 0.43 for cholesterol, P = 0.0001; r = 0.45 for triglycerides, P = 0.001).
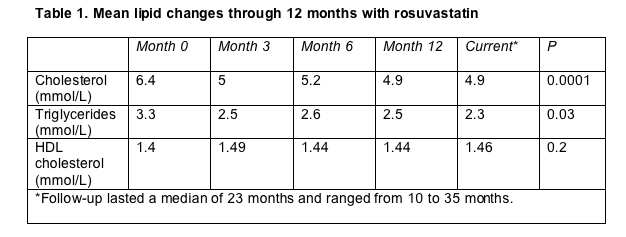
Viral load, CD4 count, hemoglobin, liver enzymes, and electrolytes did not change during follow-up. Aminotransferase levels climbed above three times normal in 1 person, who switched to pravastatin.
Atazanavir, lipids, and cardiovascular risk
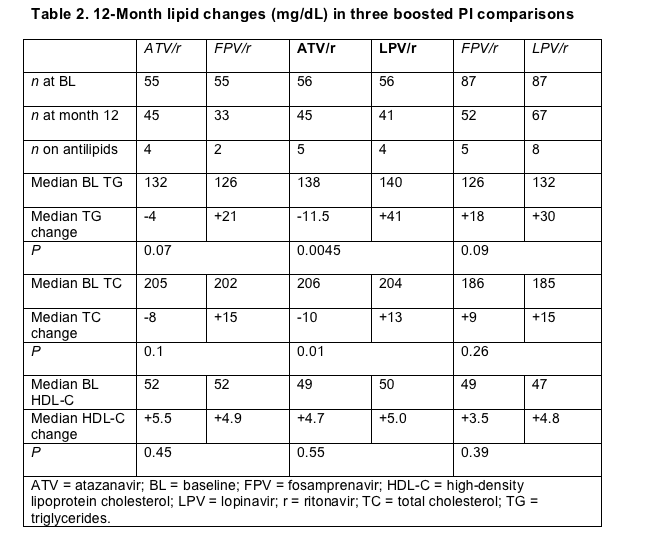
Three Glasgow studies appraised atazanavir's impact on the heart, judged by the PI's effects on lipids, endothelial function, and risk of cardiovascular disease. Stephane De Wit (Saint-Pierre University Hospital, Brussels) engineered a retrospective comparison of ritonavir-boosted atazanavir, fosamprenavir, and lopinavir by randomly selecting patients taking one of these three PIs [5]. The three groups had similar baseline lipid levels and matched closely in use of antilipid drugs (Table 2).
After 12 months of PI therapy, people taking atazanavir/ritonavir enjoyed significant drops in triglycerides and total cholesterol when compared with gains in the lopinavir/ritonavir group (Table 2). The fosamprenavir cohort also gained triglycerides and total cholesterol over 12 months of follow-up, but these gains lacked statistical significance when compared with losses in the atazanavir group (Table 2). People taking fosamprenavir did not differ significantly from those taking lopinavir in 12-month lipid changes. All three groups gained similar HDL cholesterol quotients.
A 40-patient Swiss trial that randomized people to continue their current PI or switch to atazanavir confirmed better lipid control after 12 months of atazanavir [6]. But compared with staying on current PIs, switching to atazanavir did not significantly improve endothelial dysfunction, a marker of early atherosclerosis.
Swiss clinicians at several sites recruited 40 people taking two nucleosides plus a PI for more than 12 weeks. Everyone had a viral load below 50 copies/mL and an LDL cholesterol above 3 mmol/L. The Swiss team randomized them to continue their PI or trade it for atazanavir, and study observers were not told which patients switched.
As in earlier studies, the atazanavir group had significant drops in total cholesterol (-1.23 versus -0.64 mmol/L with continued PIs, P = 0.048) and triglycerides (-1.2 versus -0.28 mmol/L, P = 0.03). LDL cholesterol (-0.89 versus -0.47, P = 0.13) and HDL cholesterol (+0.09 versus -0.012, P = 0.07) also improved with atazanavir, but those differences fell short of statistical significance. Yet 12 months of atazanavir did not significantly improve endothelial function, measured as flow-mediated vasodilation (-0.57% with atazanavir versus -0.59% with continued PIs, P = 0.95). The study was probably too small to discern a significant vasodilation difference between study arms. The Swiss team figured that each arm in this type of trial would have to include 340 people to detect a significant between-group difference.
A single-center Italian study once again confirmed the lipid-modifying benefits of switching to atazanavir, but 1 year of treatment with this lipid-friendly PI had no effect on cardiovascular risk [7]. Manuela Colafigli and colleagues at Rome's Catholic University argued that their results do support switching to atazanavir in people with out-of-line lipids, but they recommended that "intervention on other risk factors should accompany this strategy in order to obtain more significant reductions of the cardiovascular risk."
Colafigli studied 204 antiretroviral-treated patients with no history of heart disease. Three quarters of the study group smoked, 63% were men, 16% had used antihypertensive therapy, and 13% had a history of diabetes. Their age averaged 45 years and they had a median viral load of 50 copies (interquartile range [IQR] 50 to 830 copies) and a median CD4 count of 444 (IQR 305 to 595). Lipid values when they cashed in their current PI or NNRTI for atazanavir averaged 209 mg/dL for total cholesterol, 42 mg/dL for "good" HDL cholesterol, 169 mg/dL for non-HDL cholesterol, and 271 mg/dL for triglycerides.
After an average 13 months of atazanavir therapy, triglycerides fell significantly to 198.5 mg/dL (P < 0.001). Total cholesterol also slipped significantly to 191 mg/dL (P < 0.001), reflecting drops in both HDL cholesterol to 40 mg/dL (P = 0.02) and non-HDL cholesterol to 152 mg/dL (P < 0.001). Proportions of people with total cholesterol above 240 mg/dL, triglycerides above 200 mg/dL, and triglycerides above 400 mg/dL also fell during follow-up (from 23.8% to 16%, 42.8% to 35%, and 13.9% to 8%), though the proportion with non-HDL cholesterol above 190 mg/dL rose from 42.8% to 49.7%.
The Catholic University group reckoned cardiovascular risk by a validated scoring tool based on 10-year cumulative risk of a major cardiovascular "event" in an Italian population. The score does not take smoking into account, so the high smoking rate in this cohort would not drive up the risk of heart disease in this equation. But the risk calculator does factor in age, diabetes mellitus, systolic blood pressure, antihypertensive therapy, total cholesterol, and HDL cholesterol.
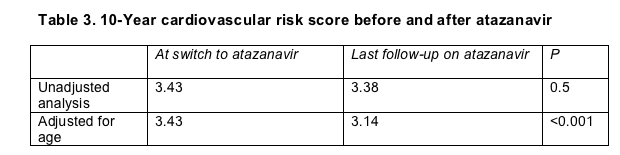
In an analysis not adjusted for other factors, the cardiovascular risk score barely budged after 13 months of atazanavir, inching down from 3.43 to 3.38 (Table 3). An analysis adjusted for age did chart a drop in heart risk score from 3.43 to 3.14. This 8% ebb reached statistical significance (P < 0.001), but Colafigli and coworkers judged it a "modest decrease." Higher total cholesterol and triglycerides before the change to atazanavir predicted bigger drops in cardiovascular risk at the end of follow-up.
Diabetes risk factors in D:A:D
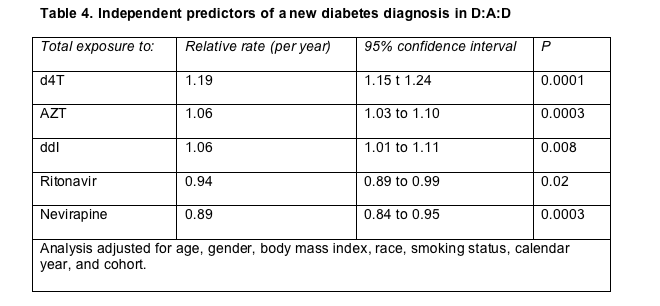
Cumulative years of treatment with stavudine (d4T) independently raised the risk of diabetes mellitus in the three-continent D:A:D cohort, as did treatment with zidovudine (AZT) and didanosine (ddI), though to a lesser extent [8]. Nevirapine and, surprisingly, ritonavir appeared to protect against diabetes, while lipodystrophy and unruly lipids raised the risk. Few physicians in D:A:D cohort countries prescribe d4T today, but the nucleoside still sees wide use in fixed-dose combinations throughout the developing world.
Stephane De Wit (Saint-Pierre University Hospital, Brussels) and colleagues sifted the 33,389-person D:A:D database looking for people with new-onset diabetes, defined as a fasting plasma glucose above 7.0 mmol/L (126 mg/dL) on two or more consecutive tests or--in people without glucose readings--the start of antidiabetes therapy (including dietary advice, insulin, or an oral antidiabetic). Including dietary advice as an antidiabetes tactic may lead to overestimation of diabetes incidence because diet may be recommended for many conditions. The research team excluded 952 people from the analysis because they already had diabetes when they entered the cohort. Of the remaining 32,437 people, 745 had a new diabetes diagnosis during 130,148 person-years of follow-up. Those numbers yielded an incidence of 5.72 diagnoses per 1000 person-years.
Statistical analysis not adjusted for other diabetes risk factors determined that the chance of getting a diabetes diagnosis rose 6% for every year of antiretroviral therapy (relative rate [RR] 1.06, 95% confidence interval [CI] 1.03 to 1.09, P = 0.0001). The rate appeared to stabilize after the first 2 to 3 years of treatment. An analysis adjusted for age, gender, body mass index, race, smoking status, calendar year, and cohort identified cumulative exposure to d4T--and to a lesser extent AZT or ddI--as independent predictors of diabetes mellitus (Table 4). Nevirapine and ritonavir appeared to lower the risk of a new diabetes diagnosis. The higher diabetes risk with d4T held true even after statistical adjustment for total cholesterol, HDL cholesterol, triglycerides, lipoatrophy, and lipohypertrophy.
The analysis confirmed several other diabetes risk factors--older age, male gender, greater body mass index, black race, injecting drug user, and earlier calendar year. But statisticians discerned no significant link between diabetes and lowest (nadir) CD4 count or duration of HIV infection upon enrollment in D:A:D.
De Wit noted that diabetes incidence in this collective cohort ran a bit lower than in other HIV cohorts, perhaps because of differences in diet and sociodemographic factors. The d4T link confirms an earlier Multicenter AIDS Cohort Study (MACS) analysis that tied this nucleoside to impaired insulin sensitivity [9].
D:A:D analysts suggested three hypotheses to explain why ritonavir--and PIs in general--did not raise the risk of diabetes, as earlier work might predict. First, use of more recent, less toxic PIs could partly explain both the calendar-year effect and the weak protective effect of ritonavir. Second, diabetes mellitus could represent the "tip of the iceberg" of upset glucose metabolism; incidence of glucose intolerance could still be higher with PIs than without them in D:A:D, but this analysis did not address glucose intolerance. Third, lipodystrophy in people with HIV may far overshadow PI therapy as a risk factor for diabetes.
Warding off abacavir hypersensitivity reactions
Routine screening for a genetic trait that predicts hypersensitivity to abacavir lowered the rate of suspected reactions from 7.5% to 3% at London's Chelsea and Westminster Hospital [10]. The finding confirms results of an earlier study of hypersensitivity reaction (HSR) screening [11].
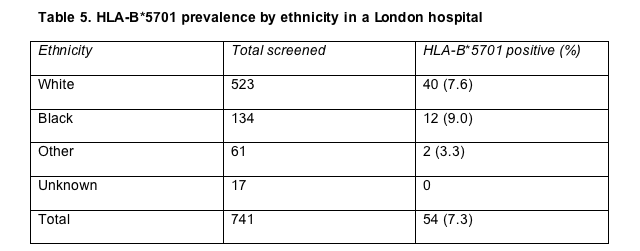
In August 2005 Chelsea and Westminster introduced routine screening for HLA-B*5701, the marker that predicts hypersensitivity [12,13], for all treatment-naive people starting antiretrovirals and all treatment-experienced people considering abacavir. Of 739 HLA-B*5701 tests done since then, Laura Waters reported, 735 were successful; 54 tests (7.3%) were positive for the abacavir HSR indicator, 9.9% in females and 6.9% in males, a nonsignificant between-gender difference.
Forty of 523 whites (7.6%) carried HLA-B*5701, as did a surprising 12 of 134 blacks (9.0%) (Table 5). As HLA-B*5701 codiscoverer Simon Mallal noted in a Glasgow talk just before Waters, the estimated prevalence of this genetic flag lies below 1% in Africa. But a second study reported in Glasgow by another London group also found a high rate of HLA-B*5701 among blacks living in Britain. Of 117 black patients tested at St. Mary's Hospital, 8 (6.8%) carried HLA-B*5701 [14]. The rate among whites was 8.8%. Christopher Collister and St. Mary's colleagues suggested sequence-based testing of the positive readings in blacks may "fully identify the *5701 allele or other family members such as *5703 . . . [and] prevent a potentially useful drug from being withheld unnecessarily." A study of 39 African and Antillean blacks in France turned up no positive tests for HLA-B*5701 [15].
Of the 54 Chelsea and Westminster patients positive for HLA-B*5701, 25 had no antiretroviral experience and 29 did. Among the 25 treatment-naive people, 14 did not begin therapy and 9 began a regimen excluding abacavir; the 2 who began abacavir both had a hypersensitivity reaction. Among the 29 treatment-experienced people with a positive test, none took abacavir after the test. Seven had taken abacavir earlier, and 4 of them had a reaction. Two of those 7 tolerated abacavir for 6 weeks and 5 years of follow-up.
Among 285 treatment-naive people negative for HLA-B*5701, 47 began abacavir and none stopped the nucleoside because of HSR. Among 396 treatment-experienced people with a negative test, 151 later switched to abacavir and 8 subsequently stopped the drug, 4 (2.6%) because of suspected HSR.
Before Chelsea and Westminster began screening for HLA-B*5701, 10 of 134 people (7.5%) starting abacavir had a suspected HSR. After screening began, 6 of 201 people (3.0%) starting abacavir had a suspected HSR (P = 0.10).
Mark Mascolini writes about HIV infection (markmascolini@earthlink.net).
References and Notes
1. Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS. 2005;19:1051-1058.
2. van der Valk M, Friis-Moller N, Sabin CA, et al. Effect of interventions to improve dyslipidaemia. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract PL9.5.
3. Between-group P values represent the results of multiple regression analyses of changes in lipids adjusted for gender, age, ethnicity, body mass index, smoking status, diabetes, previous heart disease, baseline CD4 count and viral load, number of PIs previously taken, duration of PI therapy, number of drugs taken, and current use of lopinavir, ritonavir, indinavir, or nelfinavir.
4. Das S, Boothby M, Stradling C. Rosuvastatin in the treatment of dyslipidaemia in HIV patients. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P135.
5. De Wit S, Poll B, Nescoi C, Clumeck N. Atazanavir has a better impact on lipid profiles than fosamprenavir and lopinavir in patients matched for baseline triglycerides and cholesterol. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P127.
6. Flammer A, Vo TTN, Ledergerber B, et al. Effect of atazanavir versus other protease inhibitor-containing antiretroviral combination therapy on endothelial function in HIV-infected persons: randomized controlled trial. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P95.
7. Colafigli M, Di Giambenedetto S, Bracciale L, et al. Improvement of lipid metabolism does not change the cardiovascular risk score in 13 months of observation of patients switched to atazanavir-based regimen. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P128.
8. De Wit S, Sabin CA, Weber R, et al. Relationship between use of stavudine and diabetes mellitus. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract PL9.5.
9. Brown TT, Li X, Cole SR, et al. Cumulative exposure to nucleoside analogue reverse transcriptase inhibitors is associated with insulin resistance markers in the Multicenter AIDS Cohort Study. AIDS. 2005;19:1375-1383.
10. Waters L, Gritz A, Maitland D, Nelson M. HLA-B5701 testing and abacavir hypersensitivity: a single centre experience. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract PL9.2.
11. Rauch A, Nolan D, Martin A, et al. Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study. Clin Infect Dis. 2006;43:99-102.
12. Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727-732.
13. Hetherington S, Hughes AR, Mostellar M, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359:1121-1122.
14. Collister C, Marret B, Portsmouth S, et al. HLA-B*5701 carriage frequency in a London cohort: will prospective screening be of use in our population? 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P82.
15. Zucman D, De Truchis P, Majerholc C, et al. Screening for HLA-B5701 in a population of HIV patients of diverse ethnic origin exposed to abacavir in France. 8th International Congress on Drug Therapy in HIV Infection. November 12-16, 2006. Glasgow. Abstract P116.
|
| |
|
|
|
|
|