 |
|
|
| |
Pharmacokinetic interaction between darunavir (TMC114), a new protease inhibitor, and the selective serotonin reuptake inhibitors (SSRIs), paroxetine and sertraline
|
| |
| |
Reported by Jules Levin
8th International Congress on Drug Therapy in HIV Infection, Glasgow, UK, 12-16 November 2006.
Sekar V,1 De Paepe E,2 De Marez T,1 De Pauw M,2 Vangeneugden T,2 Lefebvre E,1 Hoetelmans R2
1Tibotec Inc., Yardley, PA, USA; 2Tibotec BVBA, Mechelen, Belgium
Paroxetine (PAR) and sertraline (SER) are oral selective serotonin reuptake inhibitors (SSRIs) commonly used in the treatment of mood disorders. The current recommended dosages are 20-50mg qd for PAR10 and 50-200mg qd for SER.11
A potential pharmacokinetic (PK) drug-drug interaction may be expected when DRV/r and SER are co-administered, as these drugs are all essentially completely metabolised by CYP3A4. Furthermore, both RTV and DRV/r are potent inhibitors of CYP3A4.
PAR is mainly metabolised by CYP2D6, which plays a minor role in RTV metabolism, while both PAR and RTV are CYP2D6 inhibitors.
DRV, SER and PAR are highly bound (>95%) to plasma alpha-1-acid glycoprotein (AAG).
The primary objective of this trial was to explore the potential for PK interactions between DRV/r and SER or PAR following multiple dosing. The secondary objective was to determine short-term safety and tolerability during co-administration.
AUTHOR CONCLUSIONS
The co-administration of DRV/r with SER or PAR was generally safe and well tolerated in HIV-negative, healthy volunteers.
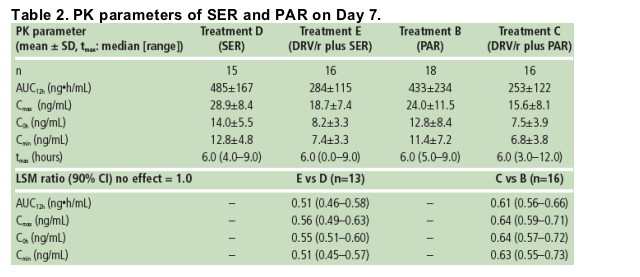
The addition of PAR or SER to a treatment regimen containing DRV/r did not affect the systemic exposure of DRV. The systemic exposure of RTV was not affected by the addition of SER to an RTV-containing regimen, but was slightly increased by the addition of PAR.
There was a decrease in exposure to SER (49%) or PAR (39%) during co-administration with DRV/r 400/100mg bid versus SER or PAR alone. Similar findings were reported in another study where PAR exposure was reduced by 55% when PAR and fosamprenavir/r were co-administered versus PAR alone.12 Unbound SER or PAR concentrations were also decreased when administered in the presence of DRV/r versus SER or PAR; the clinical relevance of these findings is unknown.
Displacement from AAG may explain, in part, the interaction between DRV and SSRIs. Modulation of transport activity may also be a potential mechanism although there is little published data suggesting that SER or PAR are substrates for transporters. Another possible explanation is induction of metabolism by RTV, although this is unlikely as SER is reportedly metabolised by different, multiple pathways.13
A comparable decrease in SER or PAR exposure is expected when SER or PAR is co-administered with the recommended DRV/r dose of 600/100mg bid.
If clinically indicated, SER or PAR may be co-administered with DRV/r. Although previous studies14,15 failed to show a correlation between SSRI concentrations and antidepressant response, clinical monitoring is recommended and dose titration of the SSRI is recommended.
Methods
Study design
This study (TMC114-C121) was a Phase I, open-label, randomised, crossover trial, conducted in two panels of 18 HIV-negative, healthy volunteers.
In three sessions, Panel 1 received Treatments A, followed by B and then C, and Panel 2 received Treatments A, followed by D and then E (all treatments were given orally as tablets or capsules under fed conditions).
Treatment A: DRV/r 400/100mg bid for 6 days with an additional morning dose on Day 7
- Treatment B: PAR 20mg qd on Days 1-7
- Treatment C: DRV/r 400/100mg bid plus PAR 20mg qd on Days 1-7
- Treatment D: SER 50mg qd on Days 1-7
- Treatment E: DRV/r 400/100mg bid plus SER 50mg qd on Days 1-7.
Subsequent sessions were separated by a washout (WO) period of at least 7 days. Volunteers stayed at the clinical unit during the treatment periods.
Pharmacokinetic analysis
PAR, SER or DRV/r with PAR or SER were administered for 7 days to allow plasma drug concentrations to reach steady-state. Predose plasma concentrations (C0h) of PAR, SER and DRV/r were determined prior to full PK sampling on Day 7 to verify that steady-state was reached.
Full PK profiles were determined for one dose interval after the morning dose on Day 7 of Treatments A, C, and E for DRV and RTV, of Treatment B and C for PAR, and of Treatments D and E for SER. Plasma concentrations of DRV, RTV, SER and PAR were determined using validated liquid chromatography mass spectrometry/mass spectrometry methods.
Unbound concentrations of SER and PAR were also measured using equilibrium dialysis at 4, 5, 6 and 9 hours post-dose for PAR, SER or DRV/r with PAR or SER treatments. Safety and tolerability were also assessed.
The least square means (LSM) of the primary PK parameters for each treatment group were estimated with a linear mixed effects model, controlling for treatment, sequence, period and individual. LSM ratios and their 90% confidence intervals (CIs) were also calculated. Period effects were considered significant at the 5% level and sequence effects were considered significant at the 10% level. If
period and sequence effects were not significant, they were removed from the model. Time to maximum plasma concentration (tmax) values were compared using the non-parametric Koch test.
Results
Volunteer disposition and baseline characteristics
Of the 36 randomised volunteers, 31 completed the trial; five volunteers discontinued due to adverse events (AEs). The median age of Panel 1 volunteers was 26 years (range 18-47 years), and of Panel 2 was 22 years (range 18-47 years); 72% of Panel 1 and 83% of Panel 2 were male.
Pharmacokinetics of DRV and RTV
Individual predose plasma concentrations of DRV and RTV determined on Days 5, 6 and 7 showed steady-state conditions were reached after 5 days of treatment.
The mean plasma concentration profiles of DRV and RTV were determined at steady-state (Figure 1).
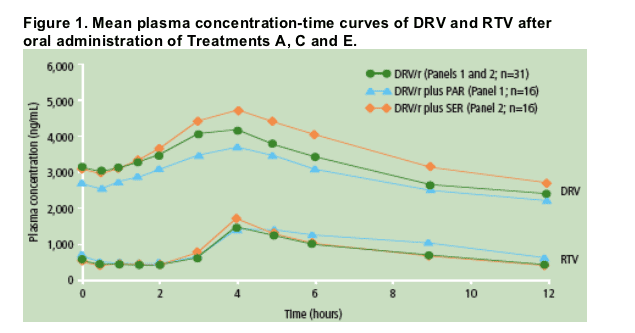
In the presence of low-dose RTV and PAR or SER, PK parameters and systemic exposure of DRV were comparable to those of treatment with DRV/r alone (Figure 1 and Table 1).
Systemic exposure of RTV in the presence of DRV and SER was comparable to that of treatment with DRV/r alone. In the presence of DRV and PAR, mean systemic exposure of RTV was approximately 34% higher compared to that of treatment with DRV/r alone (Figure 1).
Pharmacokinetics of SER and PAR
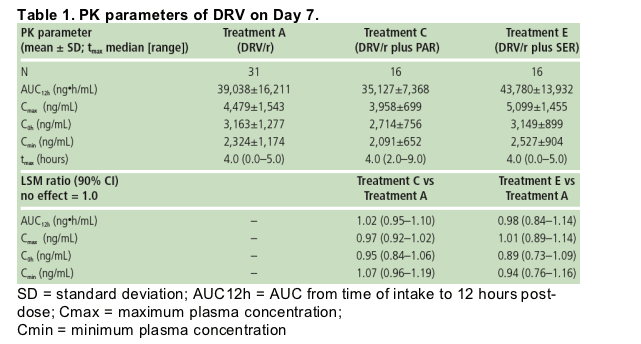
Individual predose plasma concentrations taken on Days 5, 6 and 7 showed steady-state conditions were reached within 5-6 days for SER and after 5-7 days for PAR. The mean plasma concentration profiles of SER (Figure 2) and PAR were determined at steady-state (Figure 3).
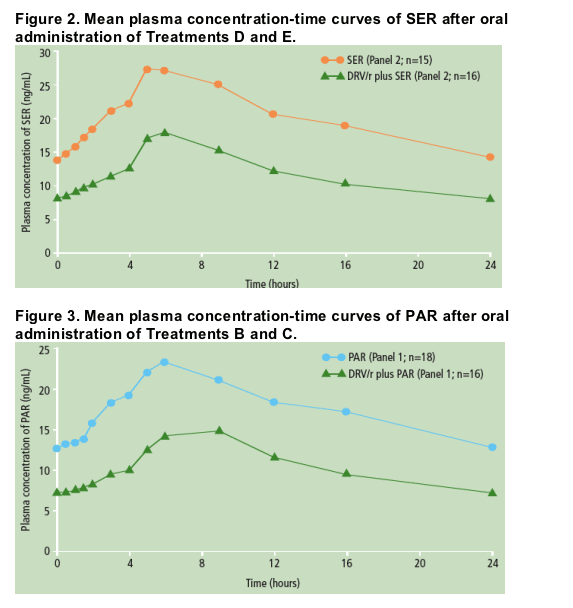
Based on the LSM ratio, there were 49% and 39% decreases in SER and PAR exposure in the presence of DRV/r compared with SER and PAR alone, respectively (Table 2).
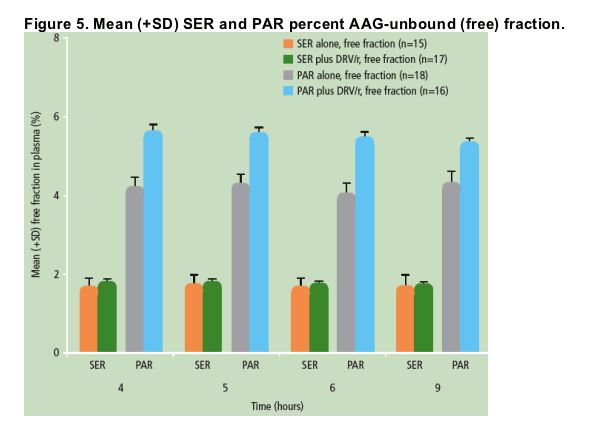
Unbound (free) concentrations of SER were decreased to a similar extent as the total concentrations of SER when SER was administered in the presence of DRV/r versus SER alone (Figure 4); the percent free fraction of SER did not change significantly between the two treatments (Figure 5).
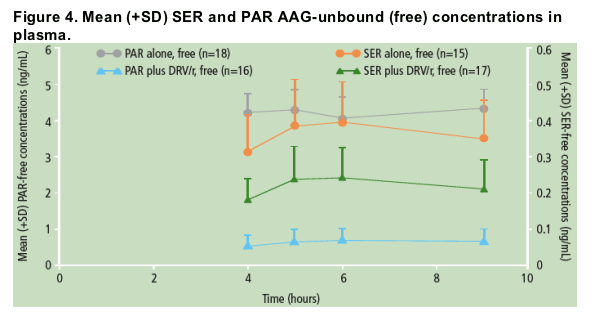
When PAR was administered in the presence of DRV/r versus PAR alone, unbound (free) concentrations of PAR were decreased, though to a lesser extent than total PAR concentrations (Figure 4); the percent free fraction of PAR was slightly increased (by 25-35%) during combined DRV/r plus PAR use versus PAR alone (Figure 5).
Safety
No serious AEs were reported.
In Panel 1, seven (41%), seven (39%) and 11 (65%) volunteers reported at least one AE during treatment with DRV/r, PAR and DRV/r plus PAR, respectively. In Panel 2, nine (64%), 11 (73%) and 13 (77%) volunteers reported at least one AE during treatment with DRV/r, SER and DRV/r plus SER, respectively; most AEs in Panel 1 or 2 were grade 1 or 2.
The most commonly reported AE was grade 1 increased blood pressure (BP); in Panel 1, this occurred for one (6%), two (11%) and three (18%) volunteers during treatment with DRV/r, PAR and DRV/r plus PAR, respectively; and in Panel 2, for two (14%), three (20%) and four (24%) volunteers during treatment with DRV/r, SER and DRV/r plus SER, respectively. None of the AEs reported was considered probably or very likely related to DRV/r.
AEs leading to discontinuation were reported for one volunteer (6%) in Panel 1 (grade 1 increased BP in WO after PAR) and for four volunteers (22%) of Panel 2 (grade 1 increased BP in WO after DRV/r/SER, grade 3 increased lipase during DRV/r/SER and for two volunteers increased liver function tests [grade 3 and grade 4 increased aspartate aminotransferase (AST) during DRV/r/SER and during SER, respectively]).
No clinically relevant treatment-related changes over time in laboratory parameters were noted. All treatment-emergent laboratory abnormalities reported during treatment phase including DRV/RTV were grade 1 or 2 in severity, except one grade 3 increase in lipase and one grade 3 increase in AST during combined DRV/r/SER treatment (reported as AEs above).
References
1. De Meyer S, et al. Antimicrob Agents Chemother 2005;49:2314-21.
2. Tibotec Inc. PREZISTATM (darunavir) Prescribing Information. June 2006 [cited 3 October 2006]. Available from: http://www.tibotectherapeutics.com/pi.jsp.
3. Katlama C, et al. 3rd IAS Conference on HIV Pathogenesis and Treatment, Rio de Janeiro, Brazil, 24-27 July 2005. Abstract WeOaLB0.102.
4. Grinsztejn B, et al. 3rd IAS Conference on HIV Pathogenesis and Treatment, Rio de Janeiro, Brazil, 24-27 July 2005. Abstract WePeLB6.201.
5. Wilkin T, et al. 45th Interscience Conference on Antimicrobial Agents and Chemotherapy,Washington DC, USA, 16-19 December 2005. Abstract 2860.
6. Berger D, et al. 45th Interscience Conference on Antimicrobial Agents and Chemotherapy,Washington DC, USA, 16-19 December 2005. Abstract H-1094.
7. Lazzarin A, et al. 16th International AIDS Conference, Toronto, Canada, 13-18 August 2006. Abstract TUAB0104.
8. Sekar V, et al. 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO, USA, 5-9 February 2006. Abstract J-121.
9. Molina JM, et al. HIV Med 2006;7(Suppl. 1):12. Abstract P4.
10. GlaxoSmithKline PAXIL® (paroxetine hydrochloride) Prescribing Information. July 2006 [cited 4 October 2006]. Available from: http://us.gsk.com/products/assets/us_paxil.pdf.
11. Pfizer Inc. ZOLOFT® (sertraline hydrochloride) Prescribing Information. July 2006 [cited 4 October 2006]. Available from: http://www.zoloft.com/pdf/ZoloftUSPI.pdf.
12. Blenke A, et al. 6th International Workshop on Clinical Pharmacology, Quebec City, Quebec, Canada, 28-30 April 2005. Abstract 13.
13. Obach RS, et al. Drug Metab and Dispos 2005;33:262-70.
14. Tasker TCG, et al. Acta Psychiatr Scand 1989;80(Suppl. 350):152-5.
15. Preskorn SH and Harvey A, Clin Pharmacol Ther 1996;59:180.
|
| |
|
|
|
|
|